Cardiac Arrest in Young Athletes
September 1, 2025
By Jacky Li, DO, and Nicolas Ellis, MD
Executive Summary
- Sudden cardiac arrest is a rare event in competitive athletes, approximately 1 in 100,000 years of participation in those aged 17 to 25 years.
- The incidence is higher in older athletes, men, Black people, and high-intensity sports, like basketball and soccer.
- The majority of cardiac arrest cases in athletes have an identifiable cause, many of which can be detected by preparticipation screening.
- Identifiable causes that can be detected by preparticipation evaluation include hypertrophic obstructive cardiomyopathy, long QT syndrome, Brugada syndrome, and arrhythmogenic right ventricular dysplasia/cardiomyopathy.
- Emergency preparedness with trained personnel and an automatic electrical defibrillator can do much to prevent death from cardiac arrest at sites of athletic training and competition.
Introduction
Sudden cardiac death (SCD) in athletes is a rare but potentially fatal event where the heart suddenly stops beating because of electrical disturbances or underlying heart conditions during physical activity.1,2 While exercise is beneficial for overall cardiovascular health, it can, in rare instances, be associated with risk of sudden death. SCD is defined by the World Health Organization as sudden, unexpected death within one hour of symptoms if the episode was witnessed or within 24 hours if unwitnessed.
As noted, exercise is a protective factor against coronary artery disease (CAD), cerebrovascular disease, and heart failure. However, factors that could increase the risk for SCD include CAD, hypertension, obesity, diabetes mellitus, cigarette smoking, and male gender. Intense physical exertion sometimes can trigger a fatal arrhythmia, such as ventricular tachycardia or fibrillation, especially in people with undiagnosed heart conditions.3 The definition of an athlete varies (gender, age, ethnicity) as does that of SCD (during exertion, after exertion, outside of exertion).
Sudden cardiac arrest in young athletes draws the public’s attention when it occurs during televised competitive events.4,5 These occurrences, although rare, may lead to an over-estimation of the risk to well-conditioned athletes by the general public. As noted later, the risk is far lower than these media reports would suggest.
Incidence
Although some professional and amateur sports leagues require mandatory reporting of injuries, there is incomplete information on the incidence of SCD in athletes.6-8 Estimates of its incidence in published reviews usually are from a mixture of incomplete databases and media reports. Starting in 1992, the U.S. National Registry of Sudden Death in Athletes was created by the Minneapolis Heart Institute Foundation to systematically gather information on SCD that occurred in young athletes participating in organized competitive sports. The National Center for Catastrophic Sport Injury Research at the University of North Carolina and the National Collegiate Athletic Association also track SCD in athletes. Analyses of data from these sources have reported incidences of SCD in athletes between 0.61 and 1.6 per 100,000 athlete participation-years and 15 in young (< 25 years of age) athletes.9-11
The incidence of SCD increases with older age, with a calculated rate of 6.64 per 100,000 person years in those older than 35 years of age.12 The observed incidence is higher in males than females and higher in competitive sports than recreational exercise.12 In the United States, the incidence of SCD is higher in Black athletes than white athletes.13,14 The incidence is higher in sports that require bursts of strenuous activity, such as basketball in the United States and football (soccer) in Europe.12
The incidence of sudden death in U.S. collegiate athletes was observed to decrease 29% by every five-year period during a 20-year study.15 This decrease was attributed to better preparticipation screening and implementation of emergency action plans with cardiopulmonary resuscitation (CPR) and automated external defibrillator (AED) resources at athletic sites.16 The conditions associated with SCD in athletes have been noted to vary according the study, likely reflecting the patient population and intensity of investigation.17
Clinical Presentation
Sudden death in athletes may be preceded by an arrhythmia associated with symptoms of palpitations, chest pain, shortness of breath, lightheadedness, dizziness, and/or presyncope or syncope during or shortly after sports activity. Athletes may notice an unexplained decline in their performance from impaired cardiac function. These symptoms indicate the need for a cardiac evaluation before resuming athletic activity.
SCD in athletes can occur without warning, requiring CPR and defibrillation. Emergency preparedness at sporting events is important to prevent death after a cardiac arrest.
The next sections of this article will discuss genetic and acquired causes associated with SCD in athletes.18
Genetic/Congenital Causes, Structurally Abnormal
Hypertrophic Obstructive Cardiomyopathy
Background/Epidemiology
Hypertrophic cardiomyopathy (HCM), particularly the obstructive form (HOCM), is a genetic heart condition characterized by abnormal thickening of the left ventricle. It is the most common cause of SCD in adolescents, with an annual SCD risk in adults ranging from 0.5% to 2%. Mutations in genes for beta-myosin heavy chain and myosin binding protein C, affecting sarcomere proteins, are common underlying causes.
Pathophysiology
The hallmark of HOCM is hypertrophy of the basal interventricular septum, leading to left ventricular outflow tract (LVOT) obstruction and left ventricular diastolic dysfunction. Symptoms, such as shortness of breath, chest pain, dizziness, and syncope during activity, can arise from this obstruction. Additionally, approximately 20% to 30% of patients experience recurrent non-sustained ventricular tachycardia or supraventricular/ventricular ectopic beats, which can degenerate into ventricular fibrillation and ultimately cause cardiac arrest. Atrial fibrillation, occurring in 2% to 3% of patients, carries a risk of thromboembolic stroke.
Diagnosis/Testing
Initial evaluation for symptomatic patients or those with suspected HOCM includes a detailed personal and family history (especially regarding SCD), physical examination, and a 12-lead electrocardiogram (ECG). Common ECG findings include left ventricular hypertrophy, ST-T segment changes, and characteristic deep, “dagger-like” Q waves. Further testing will involve formal echocardiography and, in some cases, may involve cardiac magnetic resonance imaging (CMR) or stress testing.
Bedside echocardiography by the emergency practitioner can be a useful adjunct to screen patients who present with syncope for HOCM. Under ultrasound, the maximum thickness of the ventricular wall should be assessed, with particular attention paid to the thickness of the intraventricular septum. Normal maximum wall thickness is less than 15 mm. Studies have shown that the risk of SCD from HOCM tends to increase as the thickness of the ventricular wall increases, with a maximum wall thickness greater than 30 mm a major risk factor for SCD.
Other factors associated with SCD are noted in Table 1. Restriction from competitive sports should be based on the risk of SCD.
Table 1. Risk of Sudden Death in Hypertrophic Obstructive Cardiomyopathy |
Low Risk
Moderate Risk
High Risk
|
LV = left ventricle Adapted from: Drezner JA, Malhotra A, Prutkin JM, et al. Return to play with hypertrophic cardiomyopathy: Are we moving too fast? A critical review. Br J Sports Med. 2021;55(18):1041-1047. |
Treatment
Management of HOCM involves pharmacotherapy and, in high-risk patients, implantable cardioverter/defibrillator (ICD) placement.19 Beta-adrenergic receptor blockers are the primary treatment for symptomatic patients, with L-type calcium channel blockers (e.g., verapamil, diltiazem) as alternatives. Diuretics may be used for heart failure and pulmonary congestion but require careful monitoring to avoid hypovolemia and hypotension, which can exacerbate HOCM symptoms.
Athletes who are diagnosed with HOCM should be counseled to avoid high-intensity sports, since even those athletes who are asymptomatic or minimally symptomatic carry an increased risk of SCD (up to 0.6% mortality per year), for which high-intensity exercise is a modifiable risk factor.
For patients at high risk for SCD, prophylactic ICDs are crucial, since they help terminate dangerous ventricular tachycardia/fibrillation episodes, thereby preventing cardiac arrest. Since the implementation of ICDs, HOCM-related mortality has been reduced to 0.5% per year across all age groups. However, neither ICD placement nor medical management is enough to fully reduce the risk of SCD, with 36% of HOCM patients experiencing ventricular tachycardia/ventricular fibrillation (VT/VF) even after more than 10 years with an ICD. ICD placement involves balancing benefits (arrhythmia termination, improved well-being) against risks (infection, device failure, inappropriate shocks).
While an ICD can reduce the risk of SCD in HOCM patients, the American Heart Association (AHA) and American College of Cardiology recommend that prophylactic ICDs should not be placed in athlete patients with HCM for the sole or primary purpose of permitting participation in high-intensity sports competition because of the possibility of device-related complications. Likewise, beta-blocker or ICD indications for competitive athletes with HCM should not differ from those in nonathlete patients with HCM.
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy
Background/Epidemiology
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is an autosomal dominant heart muscle disease, primarily due to gene mutations for desmosomal proteins, leading to myocyte loss and fibrofatty replacement. The estimated prevalence is 1:5,000, typically presenting between the second and fourth decades of life, with 20% presenting after 50 years of age. Initially, dysfunction predominantly affects the right ventricle, but it can progress to left-dominant or biventricular involvement, with 80% of patients having isolated right ventricular dysfunction. Progression may lead to heart failure.20
Pathophysiology
The hallmark of ARVD/C is fibrofatty replacement of right ventricular myocytes. Although it is primarily a right ventricular disease, studies show 16% to 76% of cases involve the left ventricle.21 SCD due to ARVD/C typically affects men and tends to manifest in young athletic patients during exertion. The majority of SCD cases were seen in white males, with an average age of death around 35 years.22 However, SCD also can occur because of overt heart failure from ARVD/C. Symptoms can include palpitations, lightheadedness, and syncope (a common marker for SCD risk). Associated symptoms can correlate with non-sustained/sustained ventricular arrhythmias, which may present as cardiac arrest in 19% of ARVD/C cases.
Diagnosis/Testing
Diagnosis of ARVD/C primarily is clinical, guided by the 2010 Task Force Criteria (TFC), although these criteria do not apply to dominant left ventricular types.23 Evaluation includes symptom assessment, detailed family and exercise history, ECG, ambulatory Holter monitor, echocardiography, and cardiac magnetic resonance imaging (CMR). Endomyocardial biopsies rarely are performed because of invasiveness and poor sensitivity/specificity. The most specific ECG findings are the presence of an epsilon wave in V1-V3 as well as T-wave inversions in leads V1, V2, and/or V3.23 (See Figures 1 and 2.)
Ambulatory cardiac rhythm (Holter) monitors may identify premature ventricular complexes (PVCs) and sustained/non-sustained ventricular arrhythmias.24,25 Echocardiography is the initial test for suspected ARVD/C, assessing right ventricular abnormalities, dilation, and reduced systolic function. CMR can provide detailed structural, functional, and tissue information.
Figure 1. Arrhythmogenic Right Ventricular Cardiomyopathy |
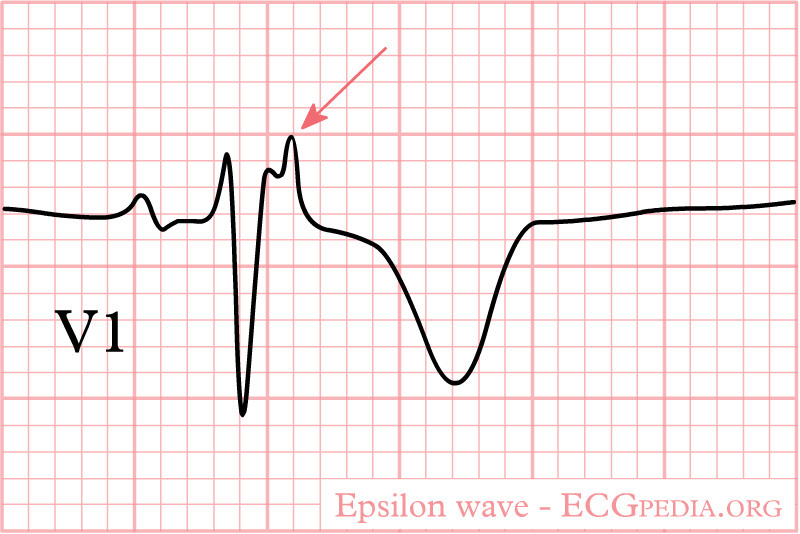 |
Source: ECGpedia. https://en.ecgpedia.org/index.php?title=Arrhythmogenic_Right_Ventricular_Cardiomyopathy &oldid=16661 |
Figure 2. Patient with Arrhythmogenic Right Ventricular Cardiomyopathy |
Patient with ARVD. T-wave inversions notable in V1 with frequent PVCs progressing to VT |
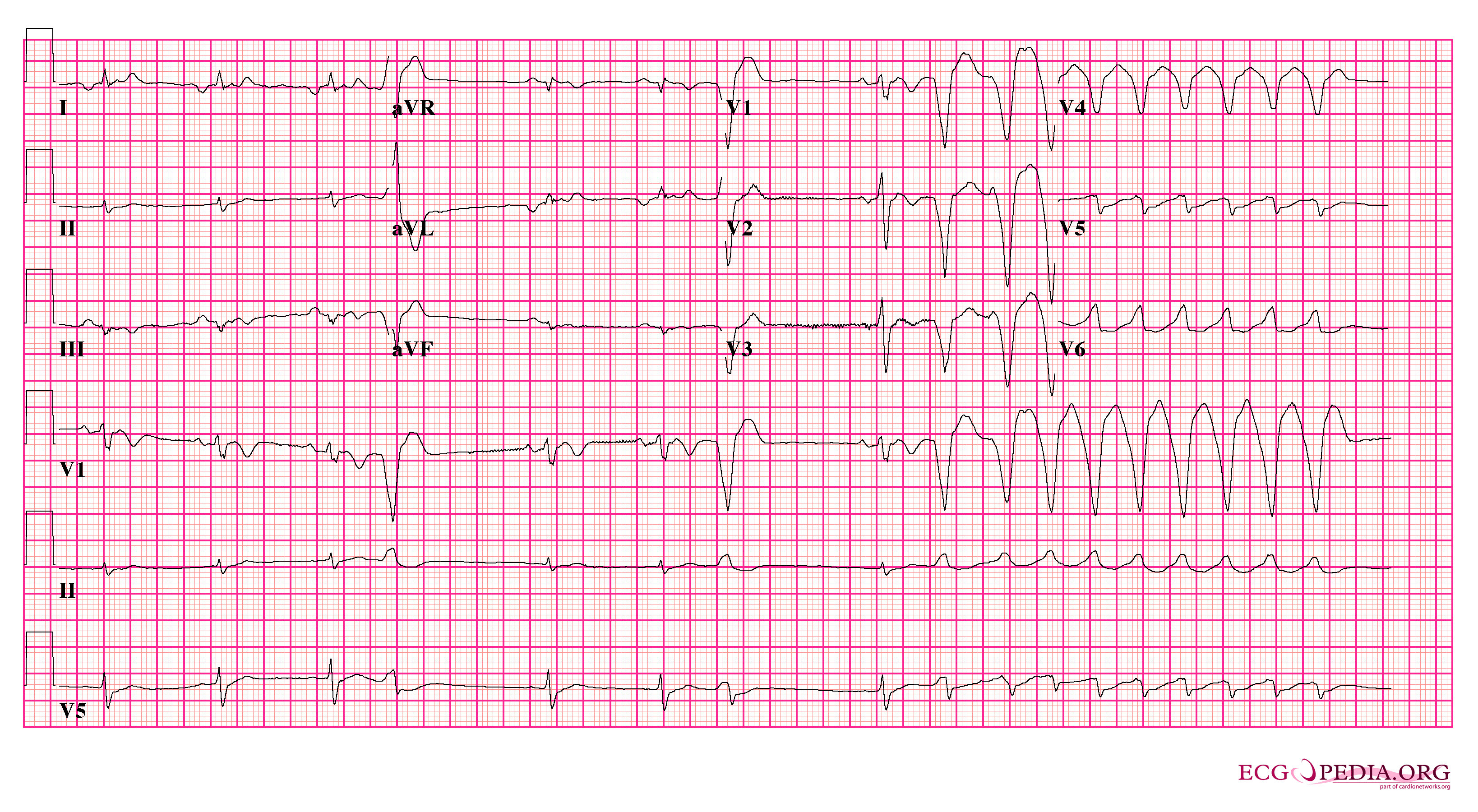 |
ARVD: arrhythmogenic right ventricular dysplasia; PVCs: premature ventricular complexes; VT: ventricular tachycardia Source: https://commons.wikimedia.org/wiki/File:Arvd_ecg2_(CardioNetworks_ECGpedia).png |
Treatment
Patients with concern for ARVD/C require cardiology consultation. The first-line pharmacologic management for ARVD/C is with beta-blockers, with anti-arrhythmics as second-line therapy (often empirically to reduce arrhythmia burden). ICD placement has significant survival benefit, since it can terminate life-threatening arrhythmias and prevent SCD, with approximately 26% survival after ICD interventions.23 Risks from ICD placement include device complications, lead complications, and inappropriate shocks.
Catheter ablation is an option for ventricular arrhythmias with frequent ICD interventions, providing short-term benefits, although recurrence of ventricular arrhythmias is seen in 50% to 70% of patients after three to five years. Exercise restriction is important, since exercise worsens ARVD/C and continued participation in competitive sports is strongly associated with increased risk for ventricular arrhythmias and SCD. Competitive sports are those that require high cardiac output, since 90% of sudden deaths in ARVD/C occur during physical exertion.
Dilated Cardiomyopathy
Background/Epidemiology
Dilated cardiomyopathy (DCM) is characterized by dilation and decreased systolic function in one or both ventricles.26,27 This impairs the heart’s pumping ability, leading to reduced cardiac output and symptoms such as fatigue, shortness of breath, and edema. DCM can be ischemic or non-ischemic, resulting from various etiologies, including genetic mutations, viral infections, alcohol use, medications, and excessive exercise.
The term “athlete’s heart” refers to dilated chambers with reduced systolic function, presumed to be caused by sustained endurance exercise.28,29 If the condition is left untreated, it can progress to heart failure or life-threatening arrhythmias, leading to SCD.25,30
DCM prevalence is approximately one in every 250 patients and carries a poor prognosis; long-term studies show approximately 50% of patients experience non-fatal life-threatening arrhythmias, unplanned cardiovascular hospitalization, or cardiovascular death over 12 years.27
Pathophysiology
Common genetic mutations in DCM involve cardiac actin, beta-myosin heavy chain, troponins, alpha-tropomyosin, and, to a lesser extent, cardiac troponin-c and myosin binding protein C. While high-intensity endurance activities can cause healthy cardiac remodeling, a small minority of elite endurance athletes develop extreme changes. Reduced left ventricular ejection fraction (LVEF), seen in approximately 16% of elite endurance athletes, is associated with a three-fold increased risk of heart failure and a 1.6-fold increased mortality risk. These severe forms of DCM can predispose young athletes to fatal arrhythmias and cardiac arrest.31
Diagnosis/Testing
DCM evaluation involves a comprehensive cardiac workup, including echocardiography, electrocardiography, and ambulatory Holter monitoring. Echocardiography is vital for initial assessment of heart muscle and signs of heart failure. DCM patients with an LVEF less than 35% have a poor prognosis. Cardiac magnetic resonance imaging is the preferred test for evaluating myocardial tissue, including dilation, edema, or fibrosis. If inflammation or inflammatory infiltrate is suspected, histology and/or immunohistology are best for assessment. An endomyocardial biopsy can identify the type of inflammatory infiltrate, although it carries complication risks.
Treatment
Patients with concern for new DCM may require admission for additional testing and cardiology consultation. Pharmacologic management of DCM focuses on treating heart failure symptoms and improving cardiac function. Primary pharmacotherapies include angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), aldosterone receptor antagonists, calcium-channel blockers, and beta-blockers (carvedilol and verapamil are particularly effective in improving LVEF).27,32
Device therapy for DCM includes cardiac resynchronization therapy (CRT), which involves cardiac pacing with either a CRT-pacemaker (CRT-P) or a combined CRT-ICD. CRT provides electrical activation of the left ventricle alone or both ventricles (biventricular pacing). It is indicated for heart failure patients with LVEF ≤ 35%, a life expectancy greater than one year, a prolonged QRS (> 130 ms), and an ECG showing left bundle branch block. CRT has been shown to reduce all-cause mortality and hospital admissions, with a relative risk ratio of death of 24% with CRT-P and 36% with CRT-ICD. For DCM patients at risk of SCD, particularly those with severely reduced LVEF, ICD implantation is a critical intervention to prevent SCD.33
Marfan Syndrome/Aortic Aneurysm Rupture
Background/Epidemiology
Marfan syndrome is an autosomal dominant connective tissue disorder caused by mutations in the extracellular matrix protein fibrillin 1.34 It affects approximately two to three individuals per 10,000 worldwide.34 The reported incidence can be influenced by factors such as increasing age of families, de-novo mutations, and lack of rapid diagnostic tests. Marfan syndrome is associated with tall stature, leading to an increased incidence in athletes such as basketball and volleyball players, and prompting screening for aortic abnormalities as noted later. A small study screening 415 basketball and volleyball athletes found aortic root dilation in four athletes and two new diagnoses of Marfan syndrome.34
Pathophysiology
Marfan syndrome affects multiple organ systems, including the cardiovascular, ocular, and skeletal systems, due to impaired connective tissue. Cardiovascular complications, particularly proximal aortic aneurysm and ascending aortic dissection, are the most common causes of SCD in Marfan syndrome.35 Because of the resultant inherent weakness of the aortic wall, activities that significantly increase blood pressure and shear stress on the aorta, such as intense athletic exertion, can precipitate acute aortic dissection or rupture, leading to catastrophic cardiac arrest.
Diagnosis/Testing
For individuals with concerns for Marfan syndrome, particularly during preparticipation screening, a detailed personal and family history is crucial. Those with a positive family history or personal risk factors should undergo echocardiography to rule out cardiac abnormalities, especially aortic root dilation.36 Major echocardiographic findings for the diagnosis of Marfan include dilation of the ascending aortic root and dissection of the ascending aorta.
Treatment
Management of Marfan syndrome involves an outpatient multidisciplinary approach focusing on prevention of complications, especially aortic rupture, which is a primary cause of SCD.37 This includes medical management (e.g., beta-blockers, ARBs to reduce aortic stress), lifestyle modifications (avoiding high-impact and contact sports and strenuous activities that increase aortic wall stress), and surgical intervention when indicated (e.g., prophylactic aortic root replacement for significant dilation). Regular monitoring of aortic dimensions with echocardiography or CMR is done to identify progression and guide timely intervention to prevent life-threatening events.
Genetic/Congenital Causes, Structurally Normal
Long QT Syndrome
Background/Epidemiology
Congenital long QT syndrome (LQTS) is a leading cause of SCD in young individuals. It is characterized by prolongation of the QT interval on the ECG and by syncopal episodes or cardiac arrest, frequently precipitated by emotional or physical stress. Without appropriate treatment, symptomatic patients face a high mortality rate, with approximately 21% mortality within one year of their first syncopal event. However, with proper management, mortality rates can be reduced to around 1% over a 15-year follow-up period.
While arrhythmic events can manifest from birth in severe cases, initial symptoms most commonly emerge around 11-12 years of age. Before puberty, females exhibit a higher risk than males. This risk equalizes between 13 and 18 years of age, after which the trend reverses.
Pathophysiology
LQTS primarily is a disorder of cardiac potassium channels. In up to 75% of cases, a genetic basis can be identified, involving mutations in genes that encode for these channels. The remaining 20% to 25% of cases lack an identifiable genetic mutation, although this does not correlate with decreased disease severity.
One well-understood subtype is LQT1, which causes mutations in the potassium channel Kv7.1. This channel generates the IKr (rapid delayed rectifier potassium current) current, crucial for appropriate QT interval adaptation during sympathetic activation and increased heart rates. When the IKr current is defective, the QT interval fails to shorten adequately during tachycardia, creating a high risk of arrhythmia. LQT1 patients are particularly vulnerable during situations that raise the heart rate, such as exercise and strong emotions, with swimming being an especially high-risk activity (99% of swimming-related arrhythmic episodes occur in LQT1 patients).
Another distinct subtype, LQT2, also is caused by potassium channel mutations and is hallmarked by exquisite sensitivity to serum potassium (K+) levels. These patients are at heightened risk when startled from sleep or rest by sudden noises. Maintaining normal serum K+ levels is critical, and potassium supplementation or K+-sparing agents may be necessary.
Gentle waking practices also are recommended for LQT2 patients to avoid sudden adrenergic surges.
Diagnosis/Testing
The hallmark of LQTS is a prolonged QT interval on the ECG.38 However, it is crucial to recognize that a normal QTc does not exclude the diagnosis, since 20% to 25% of genetically confirmed LQTS patients may have a QTc within the normal range. Furthermore, a single QTc measurement cannot definitively differentiate all LQTS from non-LQTS ECGs because of overlapping QTc values between affected individuals and healthy controls. Before attributing a prolonged QTc to LQTS, secondary causes, such as medications, acquired cardiac conditions, or electrolyte imbalances, must be meticulously excluded.
The modified Schwartz score is a tool used to calculate risk and guide the need for molecular testing (a score greater than 3 suggests a need for testing). In cases of arrhythmic syncope or cardiac arrest, a QTc ≥ 460 ms is sufficient to consider a diagnosis of LQTS.
High-risk characteristics indicative of increased severity and recurrence risk include:
- QTc > 500 ms (extremely high risk if QTc > 600 ms);
- overt T-wave electrical alternans, a direct sign of electrical instability;
- syncope or cardiac arrest before age 7 years (higher recurrence despite beta-blockers);
- syncope or cardiac arrest in the first year of life (high risk for lethal events);
- recurrent arrhythmic events despite optimal medical therapy.
Risk stratification is an important practice in this syndrome because approximately half of LQTS gene carriers never have a symptom and do not likely benefit from treatment.
Treatment
Management of LQTS can be complex.21,38 Patients who present with high-risk symptoms (syncope, cardiac arrest), and concern for LQTS requires cardiology consultation and likely admission for additional testing and monitoring.
The cornerstone of LQTS treatment is beta-blocker therapy. Non-selective beta-blockers, such as nadolol and propranolol, are the most effective agents. These medications reduce the pro-arrhythmic effects of stress and physical activity by blocking the increase in calcium current led by adrenergic stimuli, thereby preventing early afterdepolarizations (or late inward sodium current in the case of propranolol).
Beta-blocker therapy significantly reduces cardiac events and mortality, with a notable reduction in death from 60% to less than 2% per year over 10 years in symptomatic patients. LQT1 patients show the highest response rate, likely because of the antiadrenergic effect of beta-blockers on their specific trigger stimuli.
Maintaining normal serum potassium levels also is crucial, particularly in LQT2 patients, to prevent malignant arrhythmias, since the IK current is highly dependent on extracellular potassium. Potassium supplementation may be highly effective in this subtype.
For patients who have experienced cardiac arrest, an ICD is recommended as secondary prophylaxis in addition to beta-blockers (Class IB recommendation). This is because of the persistent high risk of recurrent arrhythmic events (14% within five years on therapy) in cardiac arrest survivors, even with optimal medical management.
Wolff-Parkinson-White Syndrome
Background/Epidemiology
Wolff-Parkinson-White (WPW) syndrome is a congenital cardiac conduction disorder characterized by the presence of one or more accessory pathways that bypass the atrioventricular (AV) node, predisposing patients to various arrhythmias. While it generally is considered a benign condition, it can exceptionally lead to SCD, which may be the first and only manifestation of the disease. The overall prevalence of WPW in the general population is estimated between 0.1% and 0.3% (one to three per 1,000 individuals).39
Approximately 65% of adolescents and 40% of individuals older than 30 years of age with the WPW ECG pattern are asymptomatic. The incidence of progression from a WPW pattern to symptomatic arrhythmia is thought to be around 1% to 2% per year, with the prevalence of WPW syndrome peaking between ages 20 and 24 years. The risk of SCD in WPW syndrome is low, estimated at 0.1% per year in asymptomatic patients and 0.3% per year in symptomatic patients. Population studies report SCD rates between 0.0002 to 0.0015 per patient-year for those with a WPW pattern.
Pathophysiology
The fundamental abnormality in WPW syndrome is the presence of an accessory pathway (or pathways) that provides an electrical connection between the atria and ventricles, bypassing the normal AV nodal delay. This leads to “pre-excitation,” where a portion of the ventricle is depolarized earlier than through the normal conduction system.
The most common arrhythmia associated with WPW is atrioventricular re-entrant tachycardia (AVRT), occurring in 80% of cases. Atrial fibrillation (AF) also is common, affecting one-third of patients, and is a potentially life-threatening arrhythmia in the context of WPW. Ventricular fibrillation (VF) is the most frequent cause of SCD in WPW patients.
SCD in WPW syndrome typically results from VF, often triggered by rapid conduction of atrial impulses to the ventricles via the accessory pathway. This rapid anterograde conduction can lead to extremely fast ventricular rates during atrial tachyarrhythmias (such as AF), overwhelming the ventricle and degenerating into VF.
A critical consideration is that AV nodal blocking agents (e.g., adenosine, calcium channel blockers, beta-blockers) can exacerbate this by preferentially blocking the normal AV nodal pathway, thereby shunting more impulses down the accessory pathway and increasing the ventricular rate, potentially precipitating VF.
Diagnosis/Testing
Diagnosis of WPW syndrome relies on characteristic electrocardiographic findings in conjunction with documented arrhythmia. The classic ECG features of pre-excitation include:
- a short PR interval (< 0.12 seconds);
- a “delta wave,” which is a slurring and slow rise of the initial QRS complex;
- a widened QRS complex with a total duration > 0.12 seconds;
- abnormal ventricular repolarization (secondary ST-T wave changes). (See Figure 3.)
Figure 3. Wolff-Parkinson-White Syndrome |
Ventricular pre-excitation (Wolff-Parkinson-White pattern) |
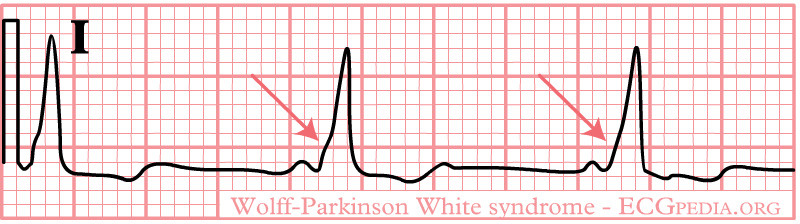 |
Source: ECGpedia. https://commons.wikimedia.org/wiki/File:De-Rhythm_WPW_(CardioNetworks_ECGpedia).png |
It is important to distinguish between a “WPW pattern” (ECG findings without symptoms) and “WPW syndrome” (ECG findings with documented arrhythmias).
Several risk factors are associated with a higher likelihood of SCD in WPW patients, including:40
- male sex;
- age younger than 35 years;
- history of atrial fibrillation or AVRT;
- presence of multiple accessory pathways;
- septal location of the accessory pathway;
- ability for rapid anterograde conduction of the accessory pathway (e.g., shortest pre-excited RR interval during AF ≤ 250 ms).41
Notably, in some studies, VF was the first manifestation of WPW syndrome in a significant proportion of patients who experienced cardiac arrest from their WPW (up to 53%), indicating that the diagnosis may be unknown prior to a cardiac arrest event. Most patients experiencing SCD with WPW do not have other associated structural heart disease. Precipitating factors for SCD in a patient with WPW include physical activity and emotional stress, both associated with catecholamine release.42
Treatment
While WPW syndrome generally is considered benign, the potential for SCD necessitates careful management. The primary goal of treatment is to prevent life-threatening arrhythmias.
A crucial aspect of management in the emergency setting is to avoid AV nodal blocking agents (e.g., adenosine, diltiazem, verapamil, beta-blockers) in patients with a WPW pattern on resting ECG who present with AF with rapid ventricular response and wide QRS complexes. These medications can paradoxically increase the risk of VF by accelerating conduction down the accessory pathway.
Although some patients may remain asymptomatic or have benign recurrence with or without medical therapy, the potential for SCD, even as a first manifestation, underscores the importance of risk stratification and consideration of invasive therapies. Definitive treatment for symptomatic WPW syndrome, particularly in those at high risk for SCD, typically involves catheter ablation of the accessory pathway.
Brugada Syndrome
Background/Epidemiology
Brugada syndrome is a distinct cardiac channelopathy characterized by a high incidence of SCD in individuals with structurally normal hearts or subtle right ventricular outflow tract abnormalities.43 It accounts for approximately 4% of all SCDs.
The prevalence of Brugada syndrome is estimated at three to five per 10,000 individuals. While it is eight to 10 times more common in males than females in the adult population (hypothesized to be because of higher post-pubertal testosterone levels and sex-specific ionic current proportions), this gender difference is not observed in pediatric patients. The syndrome also is more prevalent in individuals of Southeast Asian descent. The mean age of affected individuals is 41 years, with a wide age range from 2 days to 84 years; however, the mean age of sudden death also is 41 years. In some cases, SCD or syncope may be the first and only manifestation of the disease.44,45
Pathophysiology
The exact mechanism underlying Brugada syndrome is not fully elucidated, but it is understood to be an autosomal dominant genetic arrhythmic disease primarily affecting cardiac ion channels. Ventricular arrhythmias in Brugada syndrome commonly occur at rest, often at night and during sleep. This nocturnal predominance suggests a role for autonomic imbalance, specifically increased parasympathetic (vagal) activity and/or decreased sympathetic (adrenergic) activity in its pathophysiology. These autonomic shifts are believed to contribute to the characteristic ECG patterns and arrhythmogenesis. The lethal cardiac rhythm associated with Brugada syndrome is polymorphic ventricular tachycardia, a rapid irregular ventricular rhythm that can degenerate to ventricular fibrillation.
Diagnosis/Testing
Diagnosis of Brugada syndrome requires both a characteristic ECG finding (the “Brugada sign”) and specific clinical criteria. The ECG patterns are dynamic and can be influenced by factors such as vagal tone and certain medications. It is crucial to note that characteristic ECG morphologies recorded immediately after resuscitation or direct current (DC) shock are not diagnostic.
Three distinct ECG patterns have been described, typically observed in the right precordial leads (V1-V3):
- type 1: coved ST elevation ≥ 2 mm, followed by an inverted T wave — this is the only diagnostic pattern;
- type 2: saddleback-shaped ST elevation ≥ 2 mm;
- type 3: saddleback-shaped ST elevation < 2 mm (less clinically relevant). (See Figure 4.)
Figure 4. Brugada Syndrome |
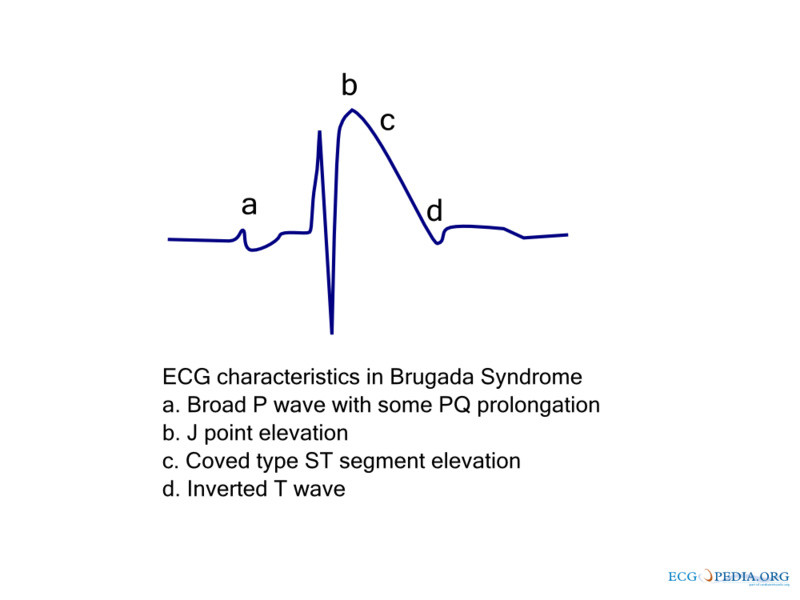 |
Source: ECGpedia. https://commons.wikimedia.org/wiki/File:De-Brugada_ecg_characteristics_(CardioNetworks_ECGpedia).png |
The diagnosis of Brugada syndrome requires the appearance of a type 1 ST-segment elevation in more than one right precordial lead (V1 to V3), either spontaneously or after administration of a sodium channel blocker, AND at least one of the following clinical criteria:
- documented VF or polymorphic ventricular tachycardia;
- family history of SCD at age < 45 years;
- coved-type ECGs in family members;
- inducibility of ventricular tachycardia with programmed electrical stimulation;
- syncope;
- nocturnal agonal respiration.
If the characteristic ECG features (Brugada sign) are present without these clinical symptoms, it is referred to as an “idiopathic Brugada ECG pattern” rather than Brugada syndrome.
Asymptomatic patterns in which the Brugada ECG pattern is found incidentally do not require consultation or follow up. Patients with a normal ECG but high-risk factors (e.g., family history of Brugada syndrome or SCD, or symptoms consistent with Brugada syndrome despite questionable ECGs) may require a provocative drug challenge test using sodium channel blockers to unmask the typical type 1 ECG pattern.
Treatment
The mainstay of treatment for Brugada syndrome patients, particularly those at high risk, is implantation of an ICD. The European Society of Cardiology (ESC) guidelines identify subjects who have survived cardiac arrest (Class I indication) and those with syncope of likely arrhythmic nature (Class IIa indication) as high-risk, for whom an ICD is clearly indicated for secondary prevention. There currently are no Class I or II indications for ICD in primary prevention.
The approach to sports participation in athletes with the Brugada ECG pattern has evolved. While historically physical activity was limited, newer studies showing a lower incidence of SCD (0.38% per year for spontaneous type 1 and 0.06% per year for pharmacologically induced type 1) have opened the possibility of competitive sports for truly low-risk patients after comprehensive screening.46 Eligibility for sports may be granted to asymptomatic individuals with type 2 or 3 patterns without a family history of juvenile SCD, or those with drug-induced type 1 without other risk factors. Eligibility may be reasonable for asymptomatic individuals with a spontaneous type 1 pattern, no family history of SCD or other minor risk factors, and a negative electrophysiological study (EPS).
Sports participation should continue to be denied for symptomatic individuals (arrhythmic syncope, resuscitated SCD) with spontaneous or drug-induced type 1 patterns, or those with a family history of SCD and a spontaneous or drug-induced type 1 pattern.
The European Association of Preventive Cardiology (EAPC) even suggests that symptomatic Brugada syndrome patients with an ICD, if asymptomatic for at least three months and observing necessary precautions, may practice all sports, including competitive ones, following a shared decision-making process with their physician. All current cardiology societies emphasize that competitive sports participation for Brugada syndrome patients is contingent on strict precautionary measures, including:
- availability of a field doctor and external defibrillator;
- avoidance of drugs that worsen the Brugada pattern;
- adequate fluid and electrolyte intake to prevent dehydration;
- prevention and timely treatment of hyperthermia (from fever or exercise);
- possession of a personal AED;
- a defined training plan and emergency action protocol with school or team officials.
Acquired — Structurally Abnormal
Atherosclerotic Coronary Artery Disease
Background/Pathophysiology
Atherosclerotic CAD occurs when plaque (fat, cholesterol) builds up in coronary arteries. This causes coronary artery narrowing and restricts blood flow to the heart muscle. SCD can result and tends to occur in athletes older than the age of 35 years. CAD is a large and widely known topic; therefore, this article will focus on its relevance to SCD in athletes.47,48
During intense physical exercise and exertion, metabolic demand increases and the heart requires more oxygen. In athletes with CAD, the presence of narrowed arteries makes it difficult for the heart to pump enough blood during exercise to meet the metabolic demands of the body. Since supply cannot meet the demand, this can lead to ischemia, arrhythmias, or myocardial infarction and ultimately SCD.47 SCD often can occur without prior warning symptoms.
Treatment/Management
Well-trained athletes may not always show signs of CAD. Risk factors for CAD include high blood pressure (hypertension), high cholesterol (hyperlipidemia), smoking tobacco, diabetes, and known family history of cardiac issues.
Early detection, prevention, and management of CAD includes regular cardiac screenings, especially in individuals older than 35 years of age and those with risk factors.49 Lifestyle changes, including exercise and diet, and medications can help reduce arterial plaque buildup.
Myocarditis
Background/Pathophysiology
Myocarditis is inflammation of the heart muscle resulting from several different etiologies.50 Causes include viral infections, bacterial infections, autoimmune disease, and certain medications. In developed nations, myocarditis typically is viral and tends to resolve spontaneously. However, in the developing world, other causes of myocarditis need to be considered, including diphtheria, rheumatic carditis, and Trypanosoma cruzi (parasite).50
Symptoms of myocarditis are nonspecific, including unexplained chest pain, dyspnea, shortness of breath, fatigue, palpitations, or syncope. Myocarditis can disrupt the heart’s electrical system and weaken the heart muscle. This can lead to arrhythmias and/or SCD.
In athletes with myocarditis, physical exercise and exertion during an infection can cause worsening of inflammation and potentially trigger fatal arrhythmias.51 SCD can occur without warning and may be a first sign of the disease.
Treatment/Management
When SCD or lethal arrhythmias occur due to myocarditis, CPR and defibrillation (AEDs) are crucial in survival, as with other causes of SCDs.52 Diagnosis of myocarditis is similar to any other cardiac workup, including ECG, echocardiography, cardiac MRI (gold standard), and tests for cardiac enzymes (blood lipids, high-sensitivity troponin, pro-B-type natriuretic peptide [BNP]) and inflammation markers (high-sensitivity C-reactive protein [CRP]).53
It is important to note that while myocarditis can be acute and short-term, individuals can develop recurrent dilated cardiomyopathy and even heart failure years later. Proper evaluation can help predict risk along with guidance for treatment.
Prevention of SCD in athletes with myocarditis includes avoiding intense physical activity during and after viral illnesses.53 Those with myocarditis are recommended to avoid exercise for three to six months, with restrictions being removed if follow-up testing is reassuring.16
Acquired Conditions — Structurally Normal
Commotio Cordis
A rare but deadly event, commotio cordis occurs when a high-energy blunt force trauma to the precordium causes rhythm disturbances leading to ventricular fibrillation.54-56 The public’s awareness of this condition was heightened by the televised sudden collapse of Damar Hamlin of the Buffalo Bills after a direct hit to the chest during an NFL game on Jan. 2, 2023.5 Commotio cordis is the second leading cause of SCD in athletes, responsible for about 20% of cases in the United States.56
The most frequent arrhythmia associated is VF, but other rhythms that can occur include polymorphic ventricular tachycardia, heart block, and AF. There are multiple factors that contribute to this, including the type of stimulus, the force of impact, the area of impact, the timing of the impact, and characteristics of the projectile.56
Commotio cordis can occur at any age, but it occurs more commonly in young males participating in contact sports. It is thought that chest wall pliability could contribute to increased risk in the younger age group, but it is unclear why males are at more risk.
In the United States alone, the leading cause of this event is baseball. (See Table 2.) Of the cases that occurred in youth baseball, they were the result of pitches thrown, on average, 30 to 50 miles per hour (mph). Interestingly, baseballs thrown less than 20 mph and between 50 and 60 mph had lower incidences of VF. The speed most likely to cause VF was 40 mph.56
Table 2. Frequency of Commotio Cordis According to Participating Sport | |
Sport | Frequency |
Baseball | 51% |
Softball | 11% |
Football | 11% |
Ice hockey | 8% |
Lacrosse | 8% |
Soccer | 6% |
Other | 5% |
Adapted from: Peng T, Derry LT, Yogeswaran V, Goldschlager NF. Commotio cordis in 2023. Sports Med. 2023;53(8):1527-1536. | |
Diagnosis/Testing
When there is a witnessed event of chest trauma, commotio cordis should be considered. This is paramount since the person could be in a dangerous arrhythmia, specifically VF, that is life-threatening. Differentials for commotio cordis include other conditions described in this article, including HOCM, ARVD, LQTS, Brugada syndrome, WPW, DCM, Marfan syndrome, CAD, and myocarditis.
Treatment
If commotio cordis is suspected and the patient is in VF, immediate treatment with early defibrillation and Advanced Cardiovascular Life Support (ACLS) protocol is critical. In an animal study, if the animal was defibrillated in less than two minutes then survivability was good. However, if they were defibrillated after four minutes, less than half survived.57 In human cases, resuscitation performed within three minutes leads to a survival rate of 25%. However, if interventions happened after three minutes, survival rates dropped to 3%.58,59
It is worth noting that the survival rate from commotio cordis in 2001 was 10% but since has increased to 15%.60 The change in survival rate likely is the result of increased awareness leading to early interventions. Since the cause of the malignant arrhythmia in commotio cordis is external and does not represent an intrinsic cardiac pathology, the primary treatment is avoidance of chest impacts, and ICD placement generally is not recommended.
Survivors of this event should have thorough cardiac evaluations that include ECG, ambulatory Holter monitoring, exercise stress test, and echocardiogram. Return to play should be reviewed on a case-by-case basis to evaluate the risk for possible event recurrence.
Inhalant Abuse: Sudden Sniffing (Death) Syndrome
Sudden sniffing syndrome (SSS) is a potentially fatal condition when an individual inhales volatile substances.61,62 These substances can include aerosol sprays, glue, paint thinner, lighter fluid, and/or household cleaning agents. These chemicals tend to be inhaled as a solvent in a container, soaked rag, or in a bag. These chemicals cause the heart to become extremely sensitive to adrenaline, which can trigger fatal heart arrhythmias including VF. Cardiac arrest after inhalation of these chemicals is unpredictable and not dose-dependent.63 Therefore, fatal cardiac events, or even death, can occur even after a single use.
SSS tends to present in young individuals in middle school and high school, with approximately 20% of children having experimented with inhalants.61 Signs and symptoms of inhalant abuse could include chemical odors, stains present on the body or clothing, behavioral changes, coordination changes leading to falls, asphyxia, burns, and frostbite.61 Chronic use can damage cardiac, renal, hepatic, and neurologic systems.
The diagnosis of SSS is difficult and mostly relies on thorough patient history taking and clinical suspicion. Treatment is supportive since there are no reversal agents for inhalants. If there is a witnessed event with immediate bystander intervention, CPR with defibrillation improves survival outcomes. Because of the sudden and deadly nature of inhalant abuse, awareness of the dangers, education, and prevention are key.
Performance-Enhancing Drugs
Performance-enhancing drugs (PEDs) are widely used by adolescents and athletes, especially bodybuilders.64,65 PEDs include anabolic steroids, stimulants, and hormones such as erythropoietin (EPO). These substances often are used to enhance performance and aesthetics, increasing muscle growth and lean body mass. While they can provide benefits, long-term and/or high-dose use can cause damage to the cardiovascular system. These substances disrupt the body’s natural balance of hormones and place significant strain on the heart. PED use has been associated with SCD.
The different types of PEDs used produce effects on the body that can lead to SCD.61 Anabolic steroids (testosterone, nandrolone, stanozolol) cause cardiac hypertrophy, commonly left ventricular hypertrophy. This leads to elevated blood pressure levels and can promote blood clot formation.
Stimulants (amphetamines, ephedrine) increase heart rate and blood pressure, which can trigger life-threatening arrhythmias. EPO can thicken blood, which increases the risk for blood clots, strokes ,and cardiac arrest. Often multiple PEDs are combined, which significantly increases the risk of adverse events.
Screening and Prevention
SCD during competitive sports is rare, but preparticipation screening can identify more than 80% of unknown cases.66-69 Prevention early on with preparticipation screenings can assess the risk of athletes before they start a sport and before intensity increases to cause symptoms.70 General preparticipation screening for multiple heart conditions and diseases includes personal and family history and physical examination. Based on current guidelines, preparticipation screening should begin whenever someone begins a competitive sport. National and local recommendations and policies will influence the age at which such screening is started. In some nations, preparticipation screening begins as early as 8 years of age.
The AHA first published recommended screening elements for preparticipation assessment of athletes in 1996. These recommendations were updated by the AHA in 2007 to include 12 total elements (eight elements of personal and family history, as well as four elements of the physical examination). In 2014, the AHA added two elements of “prior restriction from participation in sports” and “prior testing for the heart, ordered by a physician,” for the current 14-element screening.12 (See Table 3.)
Table 3. The 14-Element AHA Recommendations for Preparticipation Cardiovascular Screening of Competitive Athletes |
Medical history* |
Personal history
|
Family history
|
Physical examination
|
AHA: American Heart Association * Parental verification is recommended for high school and middle school athletes. † Judged not to be of neurocardiogenic (vasovagal origin); of particular concern when occurring during or after physical exertion. ‡ Refers to heart murmurs judged likely to be organic and unlikely to be innocent; auscultation should be performed with the patient in both the supine and standing positions (or with Valsalva maneuver), specifically to identify murmurs of dynamic left ventricular outflow tract obstruction. § Preferably taken in both arms. Reprinted with permission from: Maron BJ, Friedman RA, Kligfield P, et al. Assessment of the 12-lead ECG as a screening test for detection of cardiovascular disease in healthy general populations of young people (12-25 years of age): A Scientific Statement from the American Heart Association and the American College of Cardiology. Circulation. 2014;130:1303-1334. |
Up until this year, the AHA did not recommend the routine use of a 12-lead ECG or echocardiogram for preparticipation screening.12 The thought was that these tests were best used when initial screening raised the suspicion of cardiovascular disease. This year, a scientific statement from the AHA and American College of Cardiology stated, “The inclusion of a resting 12-lead ECG is reasonable as it improves detection of underlying cardiac conditions in asymptomatic competitive athletes compared with medical history and physical examination alone.”71 It was recognized that routine ECG use could lead to false-positive interpretations, with unjustified restrictions on sport participation and unnecessary testing and procedures. Therefore, routine ECG screening should involve clinicians trained in the interpretation of athletic ECGs paired with timely sports cardiology consultation and testing if indicated.72
Emergency Preparedness
Emergency preparedness is a critical component of athlete safety and event management in sports.73,74 Athletic events, particularly those involving high-intensity or contact activities, carry an inherent risk of acute injuries and sudden medical emergencies. A well-developed and rehearsed Emergency Action Plan (EAP) ensures that coaches, athletic trainers, medical personnel, and event staff can respond swiftly, efficiently, and in a coordinated manner when seconds matter. By anticipating potential emergencies, clearly defining roles, and ensuring the availability of essential medical equipment, event organizers can significantly reduce the risk of preventable morbidity and mortality, safeguarding both athletes and the integrity of competition.16,75-79
While this article focuses on SCD, other emergencies also can occur, including concussions, intracranial abnormalities, seizures, spinal cord injuries, respiratory emergencies, traumatic musculoskeletal injuries, and heat illnesses.80 Management for other conditions will be mentioned briefly, but detailed management can be found in other dedicated sources. It is assumed that at minimum, an initial primary survey is performed for each instance, including airway, breathing, and circulation (ABC). It also is assumed that once stabilized, the athlete will be transported by emergency medical services (EMS) to a nearby hospital for further evaluation and management of the injury.
Cardiac Emergencies/Sudden Cardiac Arrest
- Immediate response protocol: assume cardiac arrest, activate EMS, initiate CPR using the ABC approach, and deploy an AED as soon as possible.80
- The Smart Heart Sports Coalition (launched in 2023) advocates for mandated emergency plans, accessible AEDs within one to three minutes of venues, and CPR/AED training for coaches.80
Concussion/Head Injury
- Initial evaluation includes ABCs and cervical spine clearance, followed by neurological assessment.80
- Athletes with concussion symptoms must be removed from play immediately and monitored for worsening signs; reassess every 20-30 minutes during the first hour.80
Intracranial Abnormalities
- Neurologic evaluation includes Glasgow Coma Scale (GCS), orientation, pupil reactivity, extremity strength and sensation, and speech.
- Maintain awareness of red flags: loss of consciousness, worsening headache, repeated vomiting, focal neurologic deficits, seizure, or abnormal behavior.
- Management: remove from play immediately if head trauma is suspected, stabilize cervical spine, transport to hospital for evaluation.
Seizures
- Assess onset, duration, and type. Check for postictal state.
- Evaluate for head/neck trauma and any related medical history.
- Protect airway, roll to recovery position.
- If prolonged seizure, recurrent seizure, or status epilepticus, give benzodiazepine (midazolam intramuscularly [IM], lorazepam intravenously [IV]).
Spinal Cord and Cervical Injuries
- Management: assume any unconscious player has a spinal injury.80
- Stabilize the cervical spine (use spine board with minimal movement); remove facemask but leave helmet and shoulder pads in place if injury occurs during football.80
- Awareness of red flags: Similar to intracranial abnormalities but also include mechanism of injury, axial loading, limb paralysis, hypotension with bradycardia (neurogenic shock).
- Unless airway access or CPR necessitates their removal, always maintain alignment.80
Respiratory Emergencies
- These emergencies include asthma exacerbations, tension pneumothorax, and exercise-induced anaphylaxis.80
- Asthma: assess peak flow, administer short-acting beta-agonists if flow is 10% to 15% below baseline, reassess, and determine return-to-play eligibility.80
- Tension pneumothorax: recognize via breath sound loss and distress; perform needle thoracostomy if needed.80
- Anaphylaxis: watch for urticaria, angioedema, collapse; treat immediately with epinephrine, antihistamines, and steroids.80
Musculoskeletal Injuries
- Inspect for deformity, swelling, bruising, open wounds, and instability.
- Assess for tenderness, range of motion, distal pulses, capillary refill, and sensation.
- Remove from play, RICE (rest, ice, compression, elevation) therapy, immobilization, consider reduction (neurovascular compromise).
Heat Illness
- Heat illness ranges from cramps to life-threatening heat stroke.80
- Symptoms include altered mental status, seizures, nausea, and cramps. Confirm elevated core (rectal) temperature.80
- Manage heat exhaustion with cooling, rest, and hydration; treat heat stroke urgently with ice-water immersion.80
Conclusion
SCD in an athlete during physical activity has several different causes. These events can be due to underlying structural or functional abnormalities, often with preceding arrhythmias. These events can be prevented by preparticipation screenings, detailed family history investigations in families with known cardiac abnormalities, pharmacological management, and/or surgical management.
However, most abnormalities are discovered after these cardiac events occur unexpectedly. While the incidence is low, being prepared is crucial in correctly managing patients who sustain a cardiac arrest during athletic activity. Preparation readiness includes being aware of when these events occur, intervening in a timely manner, and having proper equipment available, such as AEDs.
Jacky Li, DO, is Emergency Medicine Resident, Penn State Health, Hershey, PA.
Nicolas Ellis, MD, is Associate Dean of Clinical Simulation, Assistant Professor, Emergency Medicine, Penn State College of Medicine, Hershey, PA.
References
1. Wasfy MM, Hutter AM, Weiner RB. Sudden cardiac death in athletes. Methodist Debakey Cardiovasc J. 2016;12(2):76-80.
2. Landry CH, Allan KS, Connelly KA, et al; Rescu Investigators. Sudden cardiac arrest during participation in competitive sports. N Engl J Med. 2017;377(20):1943-1953.
3. Link MS. Athletes with arrhythmias: Clinical manifestations and diagnostic evaluation. UpToDate. Last updated Jan. 18, 2024. https://www.uptodate.com/contents/athletes-with-arrhythmias-clinical-manifestations-and-diagnostic-evaluation
4. Bickel T, Gunasekaran P, Murtaza G, et al. Sudden cardiac death in famous athletes, lessons learned, heterogeneity in expert recommendations and pitfalls of contemporary screening strategies. J Atr Fibrillation. 2019;12(4):2193.
5. Kahlam J, Sacher A, Reilly JP, Lo DF. Public interest in America on cardiac arrest following cardiovascular events of Bronny and Damar: A Google trend study. Am Heart J Plus. 2024;45:100433.
6. Lear A, Patel N, Mullen C, et al. Incidence of sudden cardiac arrest and death in young athletes and military nembers: A systematic review and meta-analysis. J Athl Train. 2022;57(5):431-443.
7. Moulson N, Petek BJ, Ackerman MJ, et al. Rationale and design of the ORCCA (Outcomes Registry for Cardiac Conditions in Athletes) study. J Am Heart Assoc. 2023;12(11):e029052.
8. Tchanana GMK, Ngantcha M, Yuyun MF, et al. Incidence of recreational sports-related sudden cardiac arrest in participants over age 12 in a general African population. BMJ Open Sport Exerc Med. 2020;6(1):e000706.
9. Maron BJ, Doerer JJ, Haas TS, et al. Sudden death in young competitive athletes: Analysis of 1866 deaths in the United States, 1980-2006. Circulation. 2009;119(8):1085-1092.
10. Maron BJ, Haas TS, Murphy CJ, Ahluwalia A. Incidence and causes of sudden death in U.S. college athletes. J Am Coll Cardiol. 2014;63(16):1636-1643.
11. Harmon KG, Asif IM, Maleszewski J, et al. Incidence, cause, and comparative frequency of sudden cardiac death in national collegiate athletic association athletes: A decade in review. Circulation. 2015;132(1):10-19.
12. Han J, Lalario A, Merro E, et al. Sudden cardiac death in athletes: Facts and fallacies. J Cardiovasc Dev Dis. 2023;10(2):68.
13. Schattenkerk J, Kucera K, Peterson DF, et al. Socioeconomic factors and outcomes from exercise-related sudden cardiac arrest in high school student-athletes in the USA. Br J Sports Med. 2022;56(3):138-143.
14. Peterson DF, Kucera K, Thomas LC, et al. Aetiology and incidence of sudden cardiac arrest and death in young competitive athletes in the USA: A 4-year prospective study. Br J Sports Med. 2021;55(21):1196-1203.
15. Petek BJ, Churchill TW, Moulson N, et al. Sudden cardiac death in National Collegiate Athletic Association athletes: A 20-year study. Circulation 2024;149(2):80-90.
16. Bassi MD, Farina JM, Bombau J, et al. Sudden cardiac arrest in basketball and soccer stadiums, the role of automated external defibrillators: A review. Arrhythm Electrophysiol Rev. 2023;12:e03.
17. Finocchiaro G, Westaby J, Sheppard MN, Papadakis M. Sudden cardiac death in young athletes: JACC state-of-the-art review. J Am Coll Cardiol. 2024;83(2):350-370.
18. Kochi AN, Vettor G, Dessanai MA, et al. Sudden cardiac death in athletes: From the basics to the practical work-up. Medicina (Kaunas). 2021;57(2):168.
19. Maron MS, Rowin E, Spirito P, Maron BJ. Differing strategies for sudden death prevention in hypertrophic cardiomyopathy. Heart. 2023;109(8):589-594.
20. Kreimer F, Saguner AM, Akin I, et al. Arrhythmogenic right ventricular cardiomyopathy: Diagnosis, risk stratification, and treatment. Dtsch Arztebl Int. 2025;122(9):229-234.
21. Schnell F, Behar N, Carré F. Long-QT syndrome and competitive sports. Arrhythm Electrophysiol Rev. 2018;7(3):187-192.
22. Miles C, Finocchiaro G, Papadakis M, et al. Sudden death and left ventricular involvement in arrhythmogenic cardiomyopathy. Circulation. 2019;139(15):1786-1797.
23. Wang W, James CA, Calkins H. Diagnostic and therapeutic strategies for arrhythmogenic right ventricular dysplasia/cardiomyopathy patient. Europace. 2019;21(1):9-21.
24. Brunetti G, Cipriani A, Perazzolo Marra M, et al. Role of cardiac magnetic resonance imaging in the evaluation of athletes with premature ventricular beats. J Clin Med. 2022;11(2):426.
25. Zorzi A, De Lazzari M, Mastella G, et al. Ventricular arrhythmias in young competitive athletes: Prevalence, determinants, and underlying substrate. J Am Heart Assoc. 2018;7(12):e009171.
26. Baswaraj D, Flaker G. Syncope in athletes: A prelude to sudden cardiac death? Mo Med. 2024;121(1):52-59.
27. Schultheiss HP, Fairweather D, Caforio ALP, et al. Dilated cardiomyopathy. Nat Rev Dis Primers. 2019;5(1):32.
28. Barretta F, Mirra B, Monda E, et al. The hidden fragility in the heart of the athletes: A review of genetic biomarkers. Int J Mol Sci. 2020;21(18):6682.
29. Claessen G, De Bosscher R, Janssens K, et al; Pro@Heart Consortium. Reduced ejection fraction in elite endurance athletes: Clinical and genetic overlap with dilated cardiomyopathy. Circulation. 2024;149(18):1405-1415.
30. D’Ambrosio P, Claessen G, Kistler PM, et al. Ventricular arrhythmias in association with athletic cardiac remodelling. Europace. 2024;26(12):euae279.
31. Polovina M, Tschöpe C, Rosano G, et al. Incidence, risk assessment and prevention of sudden cardiac death in cardiomyopathies. Eur J Heart Fail. 2023;25(12):2144-2163.
32. Tong X, Shen L, Zhou X, et al. Comparative efficacy of different drugs for the treatment of dilated cardiomyopathy: A systematic review and network meta-analysis. Drugs R D. 2023;23(3):197-210.
33. Kolben Y, Hirsh Raccah B, Koev I, et al. Implantable cardioverter defibrillator for primary prevention in patients with non-ischemic cardiomyopathy in the era of novel therapeutic agents — meta-analysis. Front Cardiovasc Med. 2023;10:1192101.
34. Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501):1965-1976.
35. Hugar BS, Praveen S, Kainoor SK, Shetty AR. Sudden death in Marfan syndrome. J Forensic Sci. 2014;59(4):1126-1128.
36. Glorioso J Jr, Reeves M. Marfan syndrome: Screening for sudden death in athletes. Curr Sports Med Rep. 2002;1(2):67-74.
37. Hoffmann BA, Rybczynski M, Rostock T, et al. Prospective risk stratification of sudden cardiac death in Marfan’s syndrome. Int J Cardiol. 2013;167(6):2539-2545.
38. Gomez AT, Prutkin JM, Rao AL. Evaluation and management of athletes with long QT syndrome. Sports Health. 2016;8(6):527-535.
39. Obeyesekere MN, Leong-Sit P, Massel D, et al. Risk of arrhythmia and sudden death in patients with asymptomatic preexcitation: A meta-analysis. Circulation. 2012;125(19):2308-2315.
40. Vătășescu RG, Paja CS, Șuș I, et al. Wolf-Parkinson-White syndrome: Diagnosis, risk assessment, and therapy — An update. Diagnostics (Basel). 2024;14(3):296.
41. Leoni L, Bronzetti G, Colonna D, et al; Area Pediatrica, Associazione Italiana di Aritmologia e Cardiostimolazione (AIAC). Diagnosis and treatment of fetal and pediatric age patients (0-12 years) with Wolff-Parkinson-White syndrome and atrioventricular accessory pathways. J Cardiovasc Med (Hagerstown). 2023;24(9):589-601.
42. Vătășescu RG, Paja CS, Șuș I, et al. Wolf-Parkinson-White syndrome: Diagnosis, risk assessment, and therapy — An update. Diagnostics (Basel). 2024;14(3):296.
43. Li KHC, Lee S, Yin C, et al. Brugada syndrome: A comprehensive review of pathophysiological mechanisms and risk stratification strategies. Int J Cardiol Heart Vasc. 2020;26:100468. Erratum in: Int J Cardiol Heart Vasc. 2020;32:100699.
44. Brugada R, Campuzano O, Sarquella-Brugada G, et al. Brugada syndrome. March 31, 2005. [Updated Aug. 25, 2022]. In: Adam MP, Feldman J, Mirzaa GM, et al, editors. GeneReviews® [Internet]. University of Washington, Seattle; 1993-2025.
45. Grassi S, Campuzano O, Ferri E, et al. Forensic pathological and genetic landmarks in sudden cardiac death in the young: An update. Forensic Sci Int Genet. 2025;80:103334.
46. Gonçalves CM, Vazão A, Carvalho M, et al. Brugada syndrome and exercise: Is it time for a paradigm change? J Cardiovasc Dev Dis. 2025;12(3):94.
47. Aengevaeren VL, Eijsvogels TMH. Coronary atherosclerosis in middle-aged athletes: Current insights, burning questions, and future perspectives. Clin Cardiol. 2020;43(8):863-871.
48. Tonet E, Arzenton M, De Pietri M, et al. Coronary plaque in athletes. J Clin Med. 2024;13(7):2044.
49. Claessen G, Eijsvogels TMH, Albert CM, et al. Coronary atherosclerosis in athletes: Emerging concepts and preventive strategies. Eur Heart J. 2025;46(10):890-903.
50. Sagar S, Liu PP, Cooper LT Jr. Myocarditis. Lancet. 2012;379(9817):738-747.
51. Moulson N, Petek BJ, Baggish AL, et al. The cardiac effects of COVID-19 on young competitive athletes: Results from the Outcomes Registry for Cardiac Conditions in Athletes (ORCCA). J Cardiovasc Dev Dis. 2023;10(2):72.
52. Yan P, Yang S, Wang T. Management status of myocarditis-related sudden cardiac death. Rev Cardiovasc Med. 2024;25(12):452.
53. Bryde RE, Cooper LT Jr, Fairweather D, et al. Exercise after acute myocarditis: When and how to return to sports. Cardiol Clin. 2023;41(1):107-115.
54. Peng T, Derry LT, Yogeswaran V, Goldschlager NF. Commotio cordis in 2023. Sports Med. 2023;53(8):1527-1536.
55. Okorare O, Alugba G, Olusiji S, et al. Sudden cardiac death: An update on commotio cordis. Cureus. 2023;15(4):e38087.
56. Palacio LE, Link MS. Commotio cordis. Sports Health. 2009;1(2):174-179.
57. Link MS, Maron BJ, Stickney RE, et al. Automated external defibrillator arrhythmia detection in a model of cardiac arrest due to commotio cordis. J Cardiovasc Electrophysiol. 2003;14(1):83-87.
58. Maron BJ, Haas TS, Ahluwalia A, et al. Increasing survival rate from commotio cordis. Heart Rhythm. 2013;10(2):219-223.
59. Panhuyzen-Goedkoop NM, Wellens HJ, Verbeek ALM, et al. Immediate bystander cardiopulmonary resuscitation to sudden cardiac arrest during sports is associated with improved survival — a video analysis. Sports Med Open. 2021;7(1):50.
60. Link MS, Estes NA 3rd, Maron BJ; American Heart Association Electrocardiography and Arrhythmias Committee of Council on Clinical Cardiology, Council on Cardiovascular Disease in Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and American College of Cardiology. Eligibility and Disqualification Recommendations for Competitive Athletes With Cardiovascular Abnormalities: Task Force 13: Commotio Cordis: A Scientific Statement From the American Heart Association and American College of Cardiology. Circulation. 2015;132(22):e339-e342.
61. Anderson CE, Loomis GA. Recognition and prevention of inhalant abuse. Am Fam Physician. 2003;68(5):869-874.
62. Berling I, Isbister GK. Rare but relevant: Hydrocarbons and sudden sniffing syndrome. Addiction. 2025;120:1884-1888.
63. Tsao JH, Hu YH, How CK, et al. Atrioventricular conduction abnormality and hyperchloremic metabolic acidosis in toluene sniffing. J Formos Med Assoc. 2011;110(10):652-654.
64. Pope HG Jr, Wood RI, Rogol A, et al. Adverse health consequences of performance->enhancing drugs: An Endocrine Society scientific statement. Endocr Rev. 2014;35(3):341-375.
65. Torrisi M, Pennisi G, Russo I, et al. Sudden cardiac death in anabolic-androgenic steroid users: A Literature review. Medicina (Kaunas). 2020;56(11):587.
66. Sarto P, Zorzi A, Merlo L, et al. Value of screening for the rsik of sudden cardiac death in young competitive athletes. Eur Heart J. 2023;44(12):1084-1092.
67. Graziano F, Schiavon M, Cipriani A, et al. Causes of sudden cardiac arrest and death and the diagnostic yield of sport preparticipation screening in children. Br J Sports Med. 2024;58(5):255-260.
68. Prakash K, Swarnakari KM, Bai M, et al. Sudden cardiac arrest in athletes: A primary level of prevention. Cureus. 2022;14(10):e30517.
69. MacLachlan H, Drezner JA. Cardiac evaluation of young athletes: Time for a risk-based approach? Clin Cardiol. 2020;43(8):906-914.
70. Farzam K, Daley SF, Akhondi H. Sports participation evaluation In: StatPearls [Internet]. Updated Feb. 12, 2024. StatPearls Publishing; 2025
71. Kim JH, et al. Clinical Considerations for Competitive Sports Participation for Athletes With Cardiovascular Abnormalities: A Scientific Statement From the American Heart Association and American College of Cardiology. Circulation. 2025;151(11):e716.
72. Panhuyzen-Goedkoop NM, Wellens HJ, Verbeek AL, et al. ECG criteria for the detection of high-risk cardiovascular conditions in master athletes. Eur J Prev Cardiol. 2020;27(14):1529-1538.
73. Malik A, Hanson J, Han J, et al. Sudden cardiac arrest in athletes and strategies to optimize preparedness. Clin Cardiol. 2023;46(9):1059-1071.
74. Drezner JA, Courson RW, Roberts WO, et al. Inter-association task force recommendations on emergency preparedness and management of sudden cardiac arrest in high school and college athletic programs: A consensus statement. J Athl Train. 2007;42(1):143-158.
75. Drezner JA, Peterson DF, Siebert DM, et al. Survival after exercise-related sudden cardiac arrest in young athletes: Can we do better? Sports Health. 2019;11(1):91-98.
76. Fanous Y, Dorian P. The prevention and management of sudden cardiac arrest in athletes. CMAJ. 2019;191(28):E787-E791.
77. Vora A, Burkule N, Contractor A, Bhargava K. Prevention of sudden cardiac death in athletes, sportspersons and marathoners in India. Indian Heart J. 2018;70(1):137-145.
78. Carrington M, Providência R, Chahal CAA, et al. Cardiopulmonary resuscitation and defibrillator use in sports. Front Cardiovasc Med. 2022;9:819609.
79. Siebert DM, Drezner JA. Sudden cardiac arrest on the field of play: Turning tragedy into a survivable event. Neth Heart J. 2018;26(3):115-119.
80. Reitz L, Chambers CC. Sideline emergencies: A review of best practices. Sports Medicine Update. Fall 2023. American Orthopedic Society for Sports Medicine. https://www.sportsmed.org/membership/sports-medicine-update/fall-2023/sideline-emergencies-a-review-of-best-practices