Endocrine Causes of Secondary Hypertension
August 1, 2025
By Joy Moverley, DHSc, MSPAS, MPH, PA-C; Christopher Doan, MSPAS, MPH, PA-C; and Puja Chaudhari, PA-S, MPH(c)
Executive Summary
This issue discusses three major endocrine causes of secondary hypertension — pheochromocytoma, primary hyperaldosteronism, and Cushing syndrome — through detailed case studies and clinical guidance. It emphasizes the importance of early identification and evaluation in patients with resistant or atypical hypertension to avoid irreversible end-organ damage. The article provides diagnostic algorithms, key clinical signs, and treatment pathways to assist primary care providers in managing these conditions effectively. Enhanced awareness and timely referral are essential to optimize patient outcomes.
- Secondary hypertension should be considered in patients with resistant hypertension, early-onset hypertension before 30 years of age, sudden worsening of previously controlled blood pressure, or unexplained end-organ damage.
- Pheochromocytoma may present with episodic headaches, sweating, and tachycardia. Diagnosis involves plasma or 24-hour urinary metanephrines followed by abdominal imaging (computed tomography [CT] or magnetic resonance imaging [MRI]) for tumor localization.
- Primary hyperaldosteronism is suspected in patients with hypertension and hypokalemia. Diagnosis involves a high aldosterone-renin ratio and confirmatory sodium loading tests, with adrenal CT and adrenal venous sampling guiding treatment decisions.
- Cushing syndrome is indicated by signs such as central obesity, facial plethora, purple striae, and hirsutism, and is confirmed by at least two positive screening tests, such as overnight dexamethasone suppression, 24-hour urinary free cortisol, or late-night salivary cortisol.
- Screening for primary hyperaldosteronism is recommended in patients with difficult-to-control blood pressure, adrenal incidentalomas, hypokalemia, sleep apnea, or a family history of early cardiovascular disease.
- Surgical treatment often is curative in pheochromocytoma, unilateral hyperaldosteronism, and pituitary-driven Cushing disease; medical management is available for non-surgical candidates.
- Long-term follow-up is crucial, since residual hypertension and metabolic effects may persist even after treatment of the underlying endocrine disorder.
Endocrine-related secondary hypertension is an increasingly important area of focus as the prevalence of individuals with resistant hypertension continues to rise. Conditions such as pheochromocytomas, primary hyperaldosteronism, and Cushing syndrome are common underlying causes of secondary hypertension but often are overlooked. If not identified early enough, these disorders can rapidly accelerate an individual’s risk of developing end-organ damage and worsen prognosis.1 Despite the clinical guidelines stressing the importance of early identification to prevent such complications, healthcare professionals often are missing key diagnostic factors, resulting in underdiagnoses. The purpose of this review article is to provide clinicians with the necessary knowledge to identify these secondary causes, improving overall diagnostic accuracy and treatment outcomes.
Introduction
The 2017 American College of Cardiology (ACC) and American Heart Association (AHA) guidelines recommend initiating antihypertensive drug treatment in patients with no cardiovascular disease (CVD) risk with two separate readings of systolic blood pressure (SBP) ≥ 140 mmHg and diastolic blood pressure (DBP) ≥ 90 mmHg in people younger than 65 years of age, and SBP of ≥ 130 mmHg for those 65 years of age and older. Furthermore, the ACC/AHA define resistant hypertension as an uncontrolled office reading of blood pressure >130/80 mmHg despite the use of three or more antihypertensive medications, including a diuretic, or blood pressure of < 130/80 mmHg requiring the use of four antihypertensive medications.1 A higher level of suspicion for causes of secondary hypertension should be raised in patients with resistant hypertension. Between 5% and 10% of all adults being treated for high blood pressure have some form of resistant hypertension, thus highlighting the importance for clinicians to be well-informed and able to identify secondary hypertension, as well as its etiology.2 Moreover, up to 10% of these cases of resistant hypertension are caused by secondary hypertension.3 Identification of underlying causes is important to achieve maximal treatment potential and reduce lag time between the onset of hypertension and resolution to prevent potentially permanent end-organ damage.
When working up secondary hypertension, it is important to note that there is a broad spectrum of etiologies. Table 1 includes possible differential diagnoses for secondary causes of hypertension.4 Secondary hypertension should be considered in patients with resistant hypertension or in young-onset (younger than 30 years of age without any risk factors) hypertension, severe hypertension, sudden deterioration of blood pressure that previously was well-controlled, or presence of end-organ damage.5,6 Endocrine causes of secondary hypertension should further be suspected when there is a sudden onset, the presence of past medical history of other endocrine disorders, or an atypical presentation. When this criterion is satisfied, further investigation is warranted. This paper will focus on endocrine-related causes of secondary hypertension and will demonstrate three patient scenarios that would warrant further investigation.
Table 1. Clinical Features of Common Causes of Secondary Hypertension6 |
Renovascular Disease
Primary Renal Disease
Drug-Related Hypertension
Pheochromocytoma
Primary Aldosteronism
Cushing’s Syndrome
Sleep Apnea
Coarctation of the Aorta
Hypothyroidism
Primary Hyperparathyroidism
|
ACE: angiotensin-converting enzyme; ARB: angiotensin II receptor blocker; NSAIDs: nonsteroidal anti-inflammatory drugs; VEGF: vascular endothelial growth factor; TSH: thyroid-stimulating hormone |
Pheochromocytomas
A 52-year-old cis-gender male presents with complaints of episodic headaches, sweating, palpitations, and tremors. The symptoms have been present for multiple years but have started to become more frequent and severe in the past three months. His symptoms worsen with exercise or stress. He has a history of hypertension, for which he takes losartan, hydrochlorothiazide, and amlodipine, with no improvement in blood pressure. He denies any consumption of caffeine or illicit substances. His vital signs are body mass index (BMI), 26 kg/m²; blood pressure, 172/99 mmHg; heart rate, 120 bpm; respiratory rate, 20 breaths per minute; and temperature, 98.2°F. On the physical examination, you note an anxious-appearing adult with an elevated heart rate with a regular rhythm and no murmurs on cardiac exam. His skin is pale and diaphoretic. The initial evaluation for this patient included an electrocardiogram, which demonstrated sinus tachycardia without ectopy or ischemic changes. His laboratory tests showed the following:
- Sodium (Na): 138 mEq/L (normal range: 135 mEq/L to 146 mEq/L);
- Chloride (Cl): 102 mEq/L (normal range: 96 mEq/L to 106 mEq/L);
- Blood urea nitrogen (BUN): 19 mg/dL (normal range: 6 mg/dL to 20 mg/dL);
- Potassium (K): 3.6 mEq/L (normal range: 3.7 mEq/L to 5.2 mEq/L);
- Creatinine (Cr): 0.9 mg/dL (normal range: 0.6 mg/dL to 1.3 mg/dL);
- Glucose: 99 mg/dL (normal range: 80 mg/dL to 130 mg/dL);
- Troponin I: 0.01 ng/mL (normal range: 0 ng/mL to 0.04 ng/mL).
After the patient is evaluated to have no emergent causes of his symptoms, he undergoes further testing. An evaluation for pheochromocytomas shows elevated metanephrines.
24-hour urinary fractionated metanephrines:
- Total: 3,540 mcg/24 h (normal range: 222 mcg/24 h to 680 mcg/24 h);
- Normetanephrine: 2,240 mcg/24 h (normal range: 128 mcg/24 h to 484 mcg/24 h);
- Metanephrine: 1,300 mcg/24 h (normal range: 44 mcg/24 h to 261 mcg/24 h).
Epidemiology
Pheochromocytomas are rare endocrine tumors that develop in the adrenal medulla from chromaffin cells of the autonomic nervous system. They produce catecholamines, such as epinephrine and norepinephrine, which lead to the array of symptoms seen in patients. These tumors often are benign; however, when undiagnosed or untreated, they are associated with a high mortality rate and several complications. It is estimated that ~0.1% of people with hypertension, or two to eight people in every 1 million, have a pheochromocytoma.7
Key Symptoms and Physical Exam Findings
Pheochromocytomas can be difficult to diagnose and may be asymptomatic for many years. These tumors present with an array of symptoms, often in relation to the excess catecholamine production. Patients may have complaints of the classic clinical triad associated with pheochromocytomas, including tachycardia, headache, and excessive sweating. Other complaints may include palpitations, anxiety, and panic attacks. These patients also may present appearing pale or having sustained or paroxysmal hypertension.8
Diagnostics
Diagnosis of a pheochromocytoma relies on appropriate biochemical testing, as well as identification of a tumor on imaging.9 Blood work should be completed prior to ordering other expensive testing and imaging. Diagnostic biochemical tests in current use include measurements of urinary and plasma catecholamines, or urinary and plasma free metanephrines. Initial biochemical testing for pheochromocytomas include plasma free or urinary metanephrines.10,11 Measurements of plasma free metanephrines is more sensitive (96.6%) than urinary free metanephrines (92.9%).12 Moreover, the addition of testing for a dopamine metabolite, such as plasma 3-methoxytyramine (3-MT), increases the sensitivity (97.9%), whereas urinary 3-MT is not as favorable (93.4%).12
Measurement of plasma free normetanephrine, metanephrine, and 3-MT or 24-hour urinary fractionary metanephrines is recommended as alternative testing for the initial screening of pheochromocytoma. When measuring the 24-hour urinary excretion of fractionated metanephrines, urinary creatinine ideally should be measured to verify completeness.12
After the presence of catecholamine excess is confirmed with biochemical testing, imaging should be ordered. The optimal choice of imaging is abdominal imaging with cross-sectional contrast-enhanced computed tomography (CT); however, magnetic resonance imaging (MRI) use is acceptable in individuals who must avoid excessive radiation, such as pregnant women.10
Treatment and Prognosis
Further assessment and management of patients with pheochromocytomas ideally should be done by a multidisciplinary team with expertise in the subject. Complete tumor removal is the ultimate therapeutic goal and can be achieved by partial or total adrenalectomy with preservation of the adrenal cortex.9 Prior to surgery, patients must be managed with alpha-adrenergic blockade followed by beta-adrenergic blockade. The chronic medical management of inoperative and malignant pheochromocytomas is similar to preoperative interventions.13 Lifelong follow-up is recommended, with repeat biochemical testing as at one month, six months, and one year post-operatively. Annual follow-up is recommended thereafter. Follow-up imaging also should be repeated at one year post-operatively and repeated every one to two years.13
Primary Hyperaldosteronism
A 35-year-old cis-gender female presents with three months of periodic cramps in her legs and lethargy. She has been feeling anxious lately and sometimes feels as though her heart “skips a beat.” She was diagnosed with hypertension two months ago, and she has been given lisinopril and hydrochlorothiazide with no improvement in blood pressure. An in-office electrocardiogram was performed and showed sinus tachycardia with left ventricular hypertrophy and without ectopic beats. Her vital signs include BMI, 22 kg/m²; heart rate, 120 bpm; blood pressure, 165/92 mmHg; respiratory rate, 16 breaths per minute; and temperature, 98.8°F. On examination, the patient is a well-appearing female with a tachycardic heart rate and regular rhythm. An S4 heart sound is heard. Her laboratory tests showed the following:
- Na: 135 mEq/L (normal range: 135 mEq/L to 146 mEq/L);
- Cl: 100 mEq/L (normal range: 96 mEq/L to 106 mEq/L);
- BUN: 20 mg/dL (normal range: 6 mg/dL to 20 mg/dL);
- K: 2.9 mEq/L (normal range: 3.7 mEq/L to 5.2 mEq/L);
- Cr: 1.0 mg/dL (normal range: 0.6 mg/dL to 1.3 mg/dL);
- Glucose: 100 mg/dL (normal range: 80 mg/dL to 130 mg/dL).
The primary care provider is concerned that the patient may have primary hyperaldosteronism and orders further laboratory tests. A plasma aldosterone/renin ratio shows the following:
- Aldosterone: 33 ng/dL (normal range: 0 ng/dL to 30 ng/dL);
- Plasma renin activity: 0.9 ng/mL/hour (normal range: 0.167 ng/mL/hour to 5.380 ng/mL/hour);
- Aldosterone-renin ratio: 36.7 ng/mL/hour (normal range: 0 ng/mL/hour to 30 ng/mL/hour).
Epidemiology
Primary hyperaldosteronism, also known as Conn’s syndrome, is one common cause of secondary hypertension that potentially is curable. Primary hyperaldosteronism is characterized by the excessive production of aldosterone, resulting in increased sodium reabsorption and loss of potassium, which often leads to signs of hypernatremia, hypokalemia, and metabolic alkalosis.14
An estimated 5% to 10% of newly diagnosed hypertensive cases are caused by primary hyperaldosteronism, making primary hyperaldosteronism a leading cause of secondary hypertension.15 It also has been found to be more prevalent in patients with a low serum potassium level, those with hypertension that is resistant to one medication, and those who are older.16 In hypertensive patients with unprovoked hypokalemia, the presence of primary hyperaldosteronism reaches nearly 50%.14
Furthermore, aldosterone-producing adenomas, which are an underlying cause of primary hyperaldosteronism, are commonly seen in women younger than 40 years of age with high aldosterone levels, hypokalemia, and, often, metabolic alkalosis. However, in men, there appears to be a higher prevalence of idiopathic adrenal hyperplasia, occurring in the sixth decade of life.17
Common causes of sporadic primary hyperaldosteronism are aldosterone-producing adenoma and bilateral adrenal hyperplasia, accounting for more than 90% of all patients with primary hyperaldosteronism. The tumor often is unilateral and less than 3 cm in diameter. Aldosterone biosynthesis is more responsive to adrenocorticotropic hormone (ACTH) in patients with adenoma and more responsive to angiotensin in patients with hyperplasia. Consequently, patients with adenoma tend to have higher plasma aldosterone in the early morning that decreases during the day.14
Key Symptoms and Physical Exam Findings
Patients with primary hyperaldosteronism may present with symptoms of hypokalemia, such as fatigue, muscle weakness, headaches, hypokalemia-associated arrhythmias (such as ventricular fibrillation), and hypertension. Hypokalemia-induced nephrogenic diabetes insipidus also may occur, causing polydipsia and polyuria. Primary hyperaldosteronism should be suspected in any individual with treatment-resistant hypertension and/or hypokalemia. Many cases of primary hyperaldosteronism initially are missed, since the majority of cases have normal potassium.18 Careful diagnosis of primary hyperaldosteronism involves initial screening, confirmation of the diagnosis, and determination of a subtype.17
The age of initial diagnosis often is between 30 and 50 years. Primary hyperaldosteronism should be considered in all patients with refractory hypertension. Most patients are asymptomatic; however, there occasionally is polyuria, polydipsia, paresthesia, or muscle weakness because of hypokalemic alkalosis.14 Clinical manifestations include muscle weakness, paresthesia, polyuria, and hypertension without edema.15 The Endocrine Society recommends screening for primary hyperaldosteronism in patients with sustained blood pressure greater than 150/100 mmHg on three different measurements taken on three separate days and that is resistant to three antihypertensive medications. Table 2 demonstrates patients who should be screened for primary hyperaldosteronism.
Table 2. When to Screen for Primary Hyperaldosteronism |
Conditions that warrant screening for primary hyperaldosteronism:
|
Diagnostics
Initial screening for primary hyperaldosteronism should be done with aldosterone-renin ratio (ARR), preferably obtained in the morning. A ratio of more than 30:1 and a plasma aldosterone concentration greater than 555 pmol/L (20 ng/dL) has a sensitivity of 90% and specificity of 91% for an aldosterone-producing adenoma. If ARR is used, it is recommended that aldosterone antagonists are withdrawn for four to six weeks prior, and hypokalemia should be corrected with oral potassium supplementation prior to obtaining these laboratory tests. There also may be a falsely elevated ARR in patients with renal insufficiency as the result of decreased aldosterone clearance.14 Thus, confirmation testing should be performed.15 If confirmatory testing is positive, patients should follow up with an adrenal CT to begin the process of subtyping. Four confirmatory tests used include oral sodium loading, saline infusion, fludrocortisone suppression, and captopril challenge.15
Diagnosis and accurate treatment choices depend on good screening and subtyping of primary hyperaldosteronism. If the plasmin renin activity (PRA) is low, plasma aldosterone concentration (PAC) is high, and PAC/PRA is greater than 25, the patient should be investigated for primary hyperaldosteronism. The diagnosis of primary hyperaldosteronism is made with the demonstration of hypertension, elevated ARR, and an elevated serum aldosterone or elevated urine aldosterone excretion. If the ARR is positive, confirmatory testing should be performed to conclusively confirm or exclude the potential diagnosis. The most performed confirmatory test is salt loading, which is determined by measuring urinary aldosterone excretion. Although confirmatory testing typically is required, certain clinical scenarios allow one to bypass this step. Specifically, in cases of spontaneous hypokalemia, suppressed renin, and a PAC greater than 555 pmol/L (20 ng/dL), the diagnosis of primary hyperaldosteronism can be made without the use of further confirmatory tests.18 With confirmed primary hyperaldosteronism, the next step is to investigate the etiology of the increased serum aldosterone with an adrenal CT scan.
Bilateral adrenal venous sampling for measurement of plasma aldosterone is the most accurate means of differentiating unilateral from bilateral forms of primary aldosteronism. One frequently used protocol involves sampling for aldosterone and cortisol levels in response to ACTH stimulation. An ipsilateral/contralateral aldosterone ratio > 5 with symmetric ACTH-stimulated cortisol levels is indicative of unilateral aldosterone production.14
Treatment and Prognosis
When surgical treatment is an option for the patient, adrenal venous sampling (AVS) should be performed to determine the lateralization of aldosterone production. Individuals younger than 35 years of age with either spontaneous hypokalemia or significant aldosterone overproduction, as well as unilateral adrenal lesions that appear consistent with a cortical adenoma on CT, may be able to skip the need for AVS and proceed directly to unilateral adrenalectomy.18 For individuals with a documented unilateral primary aldosteronism, such as an aldosterone-producing adenoma or adrenal hyperplasia, unilateral adrenalectomy is recommended.18 Transient hypoaldosteronism may occur up to three months post-operatively, resulting in hyperkalemia, which should be treated with potassium-wasting diuretics, and with fludrocortisone, if needed. However, if the patient is a poor surgical candidate, or if ARR is positive and the patient is not interested or able to undergo further testing, then medical therapy with mineralocorticoid receptor antagonists is considered the preferred treatment.19 Patients with bilateral adrenal disease should be treated with pharmacotherapy, such as mineralocorticoid receptor antagonists with spironolactone or eplerenone.19
With adrenal vein sampling and no lateralization, the patient should be screened for glucocorticoid remediable aldosteronism (GRA). If found, GRA should be treated with a low dose of glucocorticoids to lower ACTH levels and improve overall control of blood pressure as well as potassium levels. If blood pressure is resistant to improvement, then the addition of a mineralocorticoid receptor antagonist is recommended.18
Hypercortisolism/Cushing Syndrome
A 42-year-old cis-gender female presents with four months of lower back pain. The pain has been increasing in severity since its onset. She also endorses generalized fatigue and states she has gained 25 pounds in the past three months. She has a history of hypertension and has been taking lisinopril without improvement in blood pressure. Her vital signs are BMI, 33 kg/m²; blood pressure, 157/79 mmHg; heart rate, 96 bpm; respiratory rate, 17 breaths per minute; and temperature, 98.4°F. On examination, the patient is centrally obese with thick violaceous striae seen on her abdomen. She also has a prominent dorsocervical fat pad, facial plethora, and coarse hair along her face, chest, and back. Her laboratory tests show the following:
- Na: 143 mEq/L (normal range: 135 mEq/L to 146 mEq/L);
- Cl: 103 mEq/L (normal range: 96 mEq/L to 106 mEq/L);
- BUN: 14 mg/dL (normal range: 6 mg/dL to 20 mg/dL);
- K: 2.7 mEq/L (normal range: 3.7 mEq/L to 5.2 mEq/L);
- Cr: 1.1 mg/dL (normal range: 0.6 mg/dL to 1.3 mg/dL);
- Glucose: 148 mg/dL (normal range: 80 mg/dL to 130 mg/dL);
- Thyroid-stimulating hormone (TSH): 3.1 μU/mL (normal range: 0.4 μU/mL to 4 μU/mL).
The primary care provider highly suspects Cushing syndrome and orders a 1-mg overnight dexamethasone suppression test (DST), which shows a cortisol level of 2.0 mcg/dL (normal range, < 1.8 mcg/dL). Alternatively, some providers may prefer to order a 24-hour urinary cortisol test, which, in this case, would demonstrate a cortisol level of 281 mcg/24 h (normal range: 4 mcg/24 h to 40 mcg/24 h).
Epidemiology
Cushing syndrome is characterized by excessive production of cortisol, resulting in intravascular volume expansion and activation of the renin-angiotensin system. This results in hypertension being present in up to 80% of cases in patients with the endogenous Cushing syndrome.20,21 Although Cushing syndrome is more common in women than men, hypertension caused by Cushing syndrome has no significant difference between men and women.21 In addition, blood pressure values are not correlated to the amount of circulating cortisol. The prevalence of hypertension is similar among exogenous and endogenous causes of Cushing syndrome.
Key Symptoms and Physical Exam Findings
Symptoms of Cushing syndrome include weight gain, lethargy, central obesity, purple skin striae, hirsutism, acne, and menstrual irregularities. Hyperpigmentation can occur in ACTH-dependent Cushing (pituitary and ectopic causes). The earliest symptoms of Cushing syndrome could be new-onset glucose intolerance, hypokalemia, and treatment-resistant hypertension.20 Therefore, diagnosis of Cushing syndrome requires a certain degree of clinical suspicion.
Diagnostics
If there is sufficient suspicion for Cushing syndrome, the patient first should undergo biochemical confirmation of hypercortisolism. This can be screened through 24-hour urine-free cortisol (UFC) excretion, overnight dexamethasone suppression, or late-night serum/salivary cortisol tests.20 At least two of the three screening tests need to be positive to confirm the presence of hypercortisolism as well as to rule out any conditions that may lead to physiologic hypercortisolism.
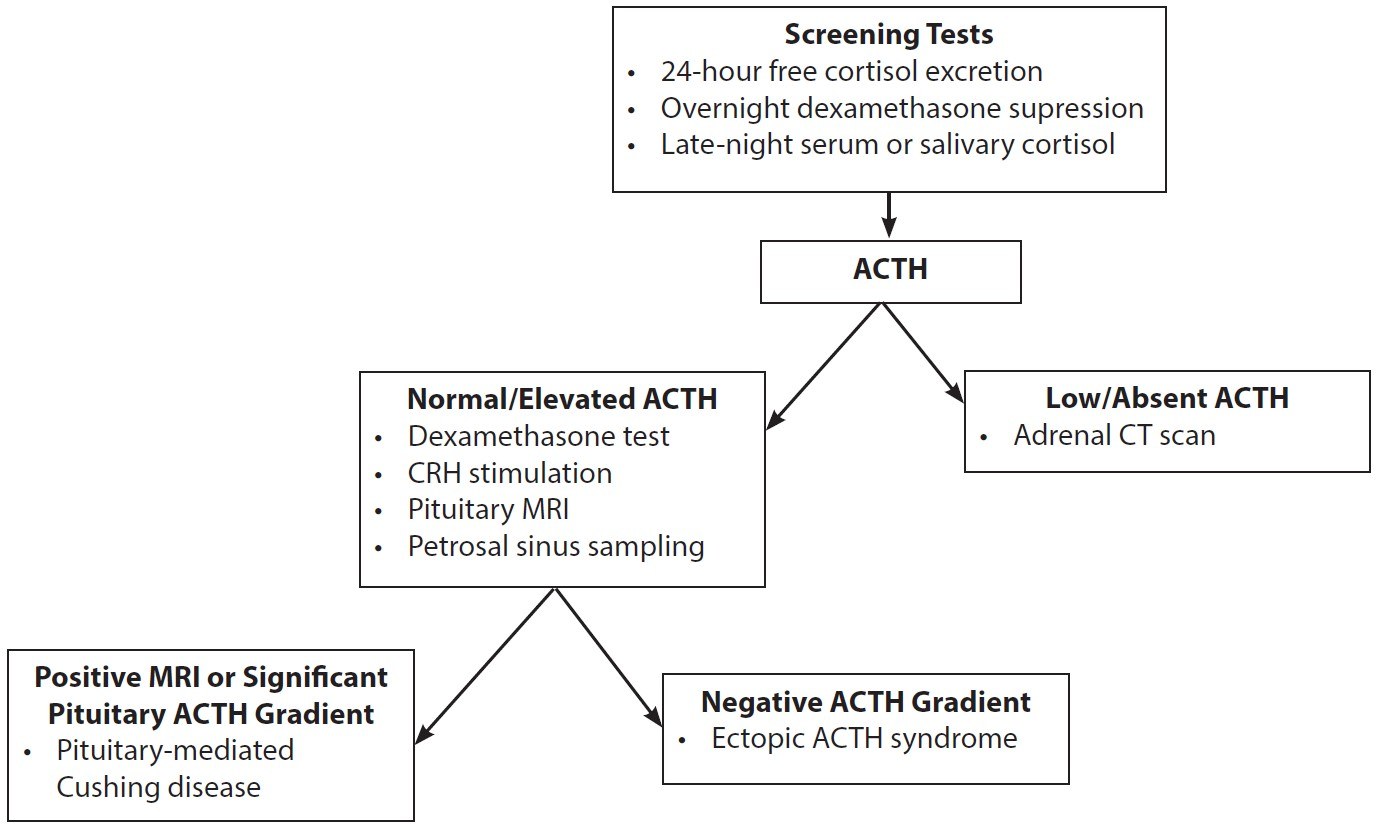
If biochemical hypercortisolism is confirmed, the next step is to determine the source of excess cortisol, which can be done through the measurement of ACTH levels. If ACTH levels are low or absent, an adrenal source of cortisol excess is suggested and prompts the use of an adrenal CT scan.20 On the other hand, if ACTH levels are normal or elevated, MRI of the pituitary is the next step. If the MRI shows a pituitary adenoma larger than 6 mm, a diagnosis of an ACTH-producing pituitary adenoma (Cushing disease) can be made.22 However, if the MRI is negative or if the adenoma is smaller than 6 mm, further testing is necessary to differentiate between a pituitary or ectopic source of ACTH. The gold standard for this differentiation is inferior petrosal sinus sampling (IPSS), which provides the most accurate results.22 Although the high-dose dexamethasone test and corticotropin-releasing hormone (CRH) stimulation test also can be used, they have lower accuracy compared to IPSS. CRH stimulation often is conducted during IPSS to enhance overall diagnostic accuracy, but it also can be used independently, although with less reliability.22
Figure 1 illustrates a stepwise manner to evaluate for Cushing syndrome vs. Cushing disease. If the MRI is positive or if there is a significant pituitary ACTH gradient, the patient has pituitary-mediated Cushing disease. If the ACTH gradient is negative, the patient has ectopic ACTH syndrome and further investigation into a potential source should be conducted.20 Table 3 summarizes the differences in laboratory findings between Cushing syndrome and Cushing disease.
Figure 1. Stepwise Manner to Evaluate for Cushing Syndrome vs. Cushing Disease |
 |
ACTH: adrenocorticotropic hormone; CRH: corticotropin-releasing hormone; MRI: magnetic resonance imaging; CT: computed tomography |
Table 3. Test Interpretation for Cushing Syndrome vs. Cushing Disease |
Cushing Syndrome 24-hour UFC: > 50 mcg/24 h ACTH: < 5 pg/mL Adrenal CT: Positive for adenomas in endogenous Cushing syndrome Cushing Disease (Pituitary) 24-hour UFC: > 50 mcg/24 h ACTH: > 20 pg/mL Pituitary MRI: Positive for pituitary adenoma (often microadenoma, negative in up to 50% of cases) Ectopic ACTH Syndrome 24-hour UFC: > 50 mcg/24 h ACTH: > 20 pg/mL (produced by ectopic tumor) Pituitary MRI: Negative Adrenal Cushing Syndrome 24-hour UFC: Often negative or < 50 mcg/24 h ACTH: Low or undetectable Adrenal CT: Positive for adrenal adenomas or tumors Pituitary MRI: Negative, unless primary adrenal tumor suspected |
ACTH: adrenocorticotropic hormone; UFC: urinary free cortisol; CT: computed tomography; MRI: magnetic resonance imaging |
Treatment and Prognosis
The primary treatment of Cushing syndrome generally is surgical intervention to address the primary cause of the increased cortisol production.20 In the case of Cushing disease, transsphenoidal selective adenectomy (TSS) is considered the optimal approach.23 When possible, surgical resection is recommended for ectopic ACTH syndrome or adrenal tumors. In cases where the patient is not a surgical candidate or if the procedure has a low probability of reducing glucocorticoid excess, medical treatments with ketoconazole are an alternative.20
Regarding comorbidities, 33% of cases of endogenous Cushing syndrome will have persistent systolic hypertension and 75% of cases of endogenous Cushing syndrome will have persistent diastolic hypertension.20 In general, treatment of Cushing syndrome results in amelioration of hypertension. However, residual hypertension persists in one-third of Cushing syndrome cases.19 Antihypertensive medications may be required in patients who continue to experience elevated blood pressure even after treatment of the underlying hypercortisolism.
The presence of persistent comorbidities and cardiovascular risk factors after disease remission serve as predictors of increased mortality, emphasizing the importance of early screening and treatment. In patients who have undergone surgical treatment, further evaluation of pituitary function is warranted to assess for any potential hypopituitarism or other hormone deficiencies.23
Conclusion
Being able to identify endocrine causes of secondary hypertension will help prevent delays of diagnosis and treatment. Three endocrine-related causes of secondary hypertension include pheochromocytomas, primary hyperaldosteronism, and Cushing syndrome.
Pheochromocytomas should be suspected in the presence of elevated metanephrines using a urinary or serum metanephrine level. Diagnosis of pheochromocytomas should include imaging modalities to confirm the presence of the tumor. Patients typically will present with the classic clinical triad of tachycardia, headaches, and diaphoresis in addition to sustained or paroxysmal hypertension.
Primary hyperaldosteronism is defined by an ARR greater than 25 and should be considered in a hypertensive patient with spontaneous or diuretic-induced hypokalemia. Patients often are well-appearing but can have evidence of left ventricular hypertrophy secondary to increased afterload from excess aldosterone. In severe cases of hypokalemia associated with primary hyperaldosteronism, ventricular fibrillation can occur.
Hypercortisolism or Cushing syndrome is characterized by elevated cortisol levels. Primary care providers should have a high level of suspicion for hypercortisolism in patients with hyperglycemia and hypertension despite treatment in addition to the classic findings of cushingoid appearance.
When managing resistant hypertension, providers should carefully consider endocrine causes in their differential diagnosis. Appropriate treatment and referrals are necessary to ensure timely management.
Joy Moverley, DHSc, MSPAS, MPH, PA-C, is Program Director, Touro University California Joint MSPAS/MPH Program, Vallejo, CA.
Christopher Doan, MSPAS, MPH, PA-C, is a Family Medicine PA, West Point Medical Center, Fontana, CA; Graduate, Touro University California Joint MSPAS/MPH Program, Vallejo, CA.
Puja Chaudhari, PA-S, MPH(c), is a Third-Year Student, Touro University California Joint MSPAS/MPH Program, Vallejo, CA.
References
1. Whelton PK, Carey RM, Aronow WS, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Hypertension. 2018;71(6):1269-1324.
2. Hegde S, Ahmed I, Aeddula NR. Secondary Hypertension. In: StatPearls. StatPearls Publishing. Last update July 20, 2023. https://www.ncbi.nlm.nih.gov/books/NBK544305/
3. Bachinsky A, Jones E, Thompson T, et al. Understanding resistant hypertension. JAAPA. 2021;34(12):15-20.
4. Textor S. Clinical features of several causes of secondary hypertension. UpToDate. Published April 15, 2022.
5. Acelajado MC, Hughes ZH, Oparil S, Calhoun DA. Treatment of resistant and refractory hypertension. Circ Res. 2019;124(7):1061-1070.
6. Hinton TC, Adams ZH, Baker RP, et al. Investigation and treatment of high blood pressure in young people: Too much medicine or appropriate risk reduction? Hypertension. 2020;75(1):16-22.
7. Aygun N, Uludag M. Pheochromocytoma and paraganglioma: From epidemiology to clinical findings. Sisli Etfal Hastanesi Tip Bul. 2020;54(2):159-168.
8. Kim JH, Moon H, Noh J, et al. Epidemiology and prognosis of pheochromocytoma/paraganglioma in Korea: A nationwide study based on the National Health Insurance Service. Endocrinol Metab (Seoul). 2020;35(1):157-164.
9. Neumann HH. Pheochromocytoma. In: Jameson J, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine, 20e. McGraw Hill; 2018.
10. Sbardella E, Grossman AB. Pheochromocytoma: An approach to diagnosis. Best Pract Res Clin Endocrinol Metab. 2020;34(2):101346.
11. Shen Y, Cheng L. Biochemical diagnosis of pheochromocytoma and paraganglioma. In: Mariani-Costantini R, ed. Paraganglioma: A Multidisciplinary Approach [Internet]. Codon Publications; 2019.
12. Eisenhofer G, Prejbisz A, Peitzsch M, et al. Biochemical diagnosis of chromaffin cell tumors in patients at high and low risk of disease: Plasma versus urinary free or deconjugated O-methylated catecholamine metabolites. Clin Chem. 2018;64(11):1646-1656.
13. Farrugia FA, Charalampopoulos A. Pheochromocytoma. Endocr Regul. 2019;53(3):191-212.
14. Kotchen TA. Hypertensive vascular disease. In: Jameson J, Fauci AS, Kasper DL, et al, eds. Harrison’s Principles of Internal Medicine, 20e. McGraw Hill; 2018.
15. Tezuka Y, Yamazaki Y, Nakamura Y, et al. Recent development toward the next clinical practice of primary aldosteronism: A literature review. Biomedicines. 2021;9(3):310.
16. Zilbermint M, Hannah-Shmouni F, Stratakis CA. Genetics of hypertension in African Americans and others of African descent. Int J Mol Sci. 2019;20(5):1081.
17. Cobb A, Leslie SW. Primary Hyperaldosteronism. In: StatPearls. StatPearls Publishing. Last update May 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK539779/
18. Funder JW, Carey RM, Mantero F, et al. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2016;101(5):1889-1916.
19. Schernthaner-Reiter MH, Siess C, Gessl A, et al. Factors predicting long-term comorbidities in patients with Cushing’s syndrome in remission. Endocrine. 2019;64(1):157-168.
20. Torre JJ, Bloomgarden ZT, Dickey RA, et al; AACE Hypertension Task Force. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the diagnosis and treatment of hypertension. Endocr Pract. 2006;12(2):193-222. [Erratum in: Endocr Pract. 2008;14(6):802-803.]
21. Barbot M, Ceccato F, Scaroni C. The pathophysiology and treatment of hypertension in patients with Cushing’s syndrome. Front Endocrinol (Lausanne). 2019;10:321.
22. Nieman LK, Biller BMK, Findling JW, et al. The diagnosis of Cushing’s syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93(5):1526-1540.
23. Nieman LK, Biller BMK, Findling JW, et al. Treatment of Cushing’s syndrome: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2015;100(8):2807-2831.